Totální syntéza strychninu
Totální syntéza strychninu je označení pro různé posloupnosti chemických reakcí vedoucích k umělé syntéze strychninu. První takový postup popsal Robert Burns Woodward v roce 1954.[2][3][4][5]
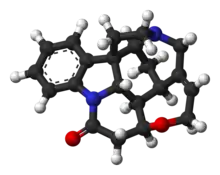
Rozvoji metod určování struktury molekul započal s izolací strychninu z kulčiby hořké (Strychnos ignatii), kterou provedli Pierre Joseph Pelletier a Joseph Bienaimé Caventou v roce 1818.[6][7] Hlavní podíl na tom měli Robert Robinson, který na toto téma vydal přes 250 článků, a Hermann Leuchs se 125 články vydanými v rozmezí 40 let. Robinson získal v roce 1947 Nobelovu cenu za chemii za výzkum alkaloidů, včetně strychninu.
Proces určování chemické struktury byl zakončen v roce 1946[8][9][10] a potvrzen roku 1947.[11] Pomocí rentgenové krystalografie byla mezi roky 1947 a 1951 určena absolutní konfigurace, o což se zasloužili Johannes Martin Bijvoet[12][13] a J. H. Robertson.[14][15]
Robert Burns Woodward vydal v roce 1954 kratší (obsahující pouze 3 strany)[16] a v roce 1963 delší (obsahující 42 stran) poznámku k syntéze strychninu.[17]
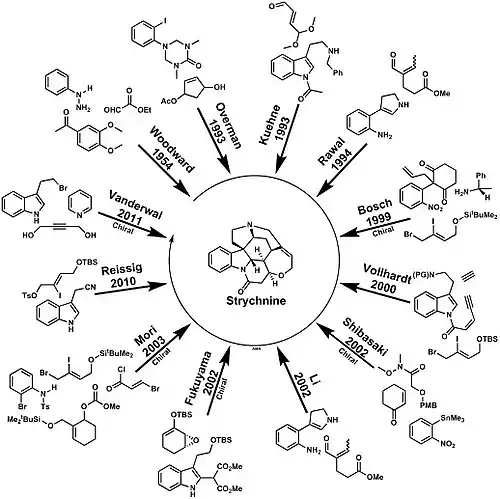
Mnoho dalších postupů popsali Philip Magnus,[18] Larry E. Overman,[19] Martin E. Kuehne,[20][21] V. H. Rawal,[22] Joan Bosch,[23][24] Peter C. Vollhardt,[25][26] Miwako Mori,[27][28] Masakatsu Shibasaki,[29] Li,[30] T. Fukujama[31] Christopher D. Vanderwal [32] a David W. C. MacMillan.[33] Je také znám syntetický (+)-strychnin.[34] Syntézy racemátu byly zveřejněny v letech 2007[35] a 2010.[36][37]
V roce 1963 citoval Woodward tvrzení Roberta Robinsona,[38] že jde o z hlediska velikosti molekuly o nejsložitější látku, která je známa.
Struktura
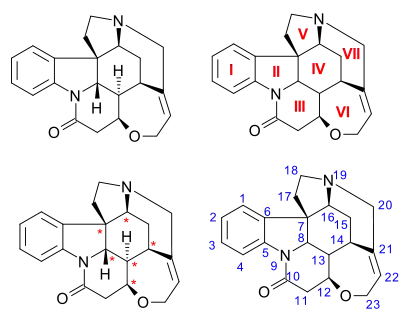
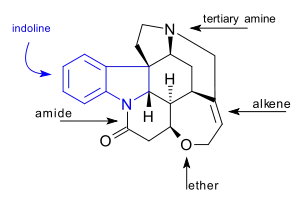
Molekula strychninu má souhrnný vzorec C21H22N2O2, se sedmi kruhy v indolinovém systému. Obsahuje terciární amin, karboxamid, alken a ether. Rovněž vykazuje chiralitu, jelikož má 6 asymetrických atomů uhlíku, jeden z nich je kvartérní.
Woodwardova syntéza
Syntéza kruhů II a V
Kruh II byl vytvořen pomocí Fischerovy syntézy indolů z fenylhydrazinu 1 a acetoveratronu, látky odvozené od acetofenonu, 2 (katalyzátorem byla kyselina polyfosforečná) za vzniku 2-veratrylindolu 3. Veratrylová skupina brání dalším elektrofilním substitucím do polohy 2 a současně se stává součástí molekuly strychninu. Mannichovou reakcí s formaldehydem a dimethylaminem se vytvořil gramin 4. Alkylací jodmethanem vznikla kvartérní amonná sůl, která podstoupila nukleofilní substitucční reakci s kyanidem sodným na nitril 5, který byl redukován tetrahydridohlinitanem lithným na tryptamin 6. amino-karbonylovou kondenzací s ethylesterem kyseliny glyoxalové za tvorby iminu 7. Reakcí tohoto iminu s tosylchloridem (TsCl) v pyridinu se vytvořila N-tosylová sloučenina 8 obsahující uzavřený řetězec. Tato sloučenina by měla tvořit diastereomerní pár, byla však zjištěna tvorba pouze jednoho diastereomeru, i když nebylo zjišťováno, kterého. Takto vzniklá dvojná vazba se zredukovala tetrahydridoboritanem sodným na indolin 9.
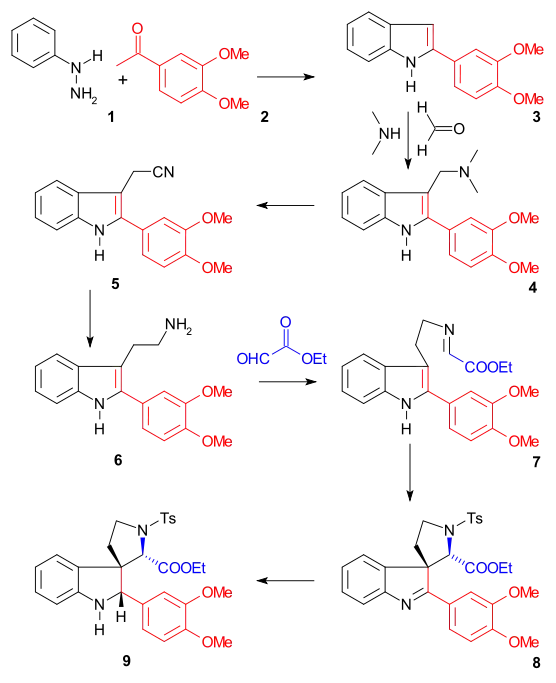
Syntéza kruhů III a IV
Indolin 9 byl acetylován za tvorby N-acetylu 10 anhydridem kyseliny octové v pyridinu, přičemž se provedlo otevření kruhu na veratrylové skupině pomocí ozonu v roztoku kyseliny octové za vzniku mukonátu 11 (umožněného methoxidovými skupinami, dodávajícími dva elektrony).[39] Odštěpením acetylu a hydrolýzou esteru kyselinou chlorovodíkovou v methanolu vznikl pyridonový ester 12 a izomerizaci exocyklické dvojné vazby na endocyklickou (přičemž zaniklo jedno asymetrické centrum). Následným působením jodovodíku a červeného fosforu byla odstraněna tosylová skupina a zbývající esterové skupiny se přeměnily na dikarboxylovou kyselinu 13. Acetylací a esterifikací v diazomethanu vznikl acetylový diester 14, jenž následně vstoupil do Dieckmanovy kondenzace s methoxidem sodným v methanolu, čímž vznikl enol 15.
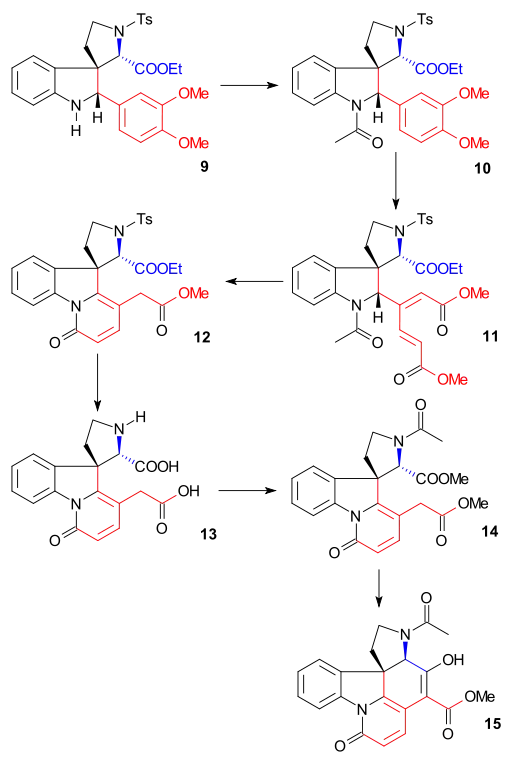
Syntéza kruhu VII
K odstranění alkoholové skupiny na C15 byl enol 15 přeměněn na tosylát 16, reakcí s tosylchloridem v pyridinu, a následně pomocí benzylthiolátu sodného převeden na thioester 17; ten byl poté redukován na nenasycený ester 18 vodíkem za přítomnosti Raneyova niklu. Další redukcí vodíkem, katalyzovanou palladiem na uhlíku se vytvořil nasycený ester 19. Alkalickou hydrolýzou tohoto esteru došlo k epimerizaci na C14.
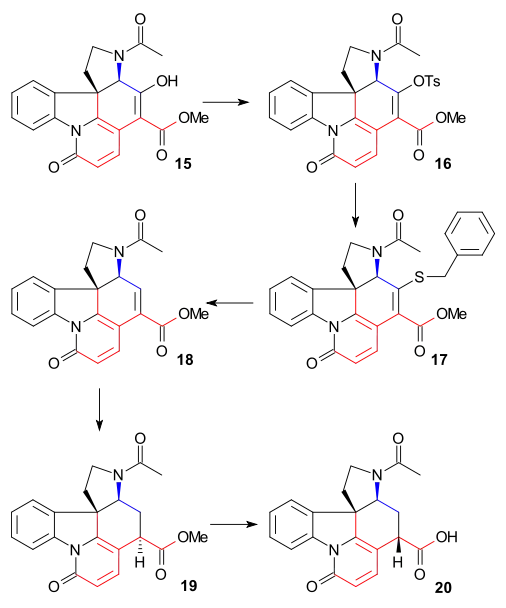
Tato sloučenina již byla popsána ve studiích rozkladu strychninu. Do tohoto okamžiku byly všechny meziprodukty racemické, zde byla chiralita zavedena chirálním rozlišením s použitím chinidinu.
Uhlík C20 se do řetězce dostal působením acetanhydridu, který vytvořil enolacetát 21. Následně se utvořil volný aminoketon 22 hydrolýzou s využitím kyseliny chlorovodíkové. Kruh VII u meziproduktu23 byl uzavřen oxidací oxidem seleničitým, což bylo opět provázeno epimerizací na C14.
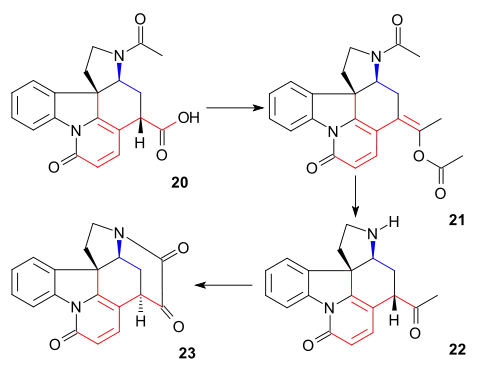
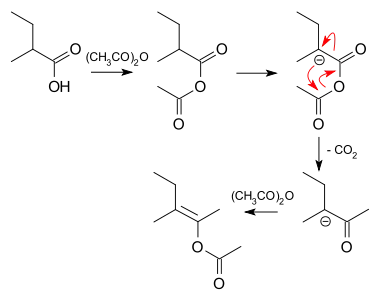
Tvorbu 21 lze provést posloupností acylace, deprotonace, přesmyku s oddělením oxidu uhličitého a opětovné acylace:
Syntéza kruhu VI
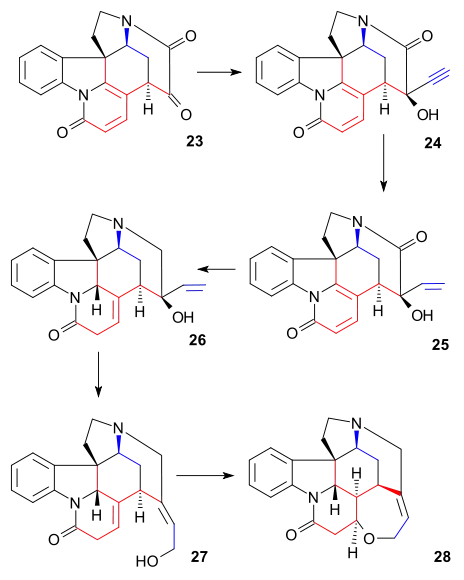
Diketon 23 reagoval s acetylidem sodným za vzniku alkynu 24. Tato sloučenina se zredukovala na alkohol 25 pomocí Lindlarova katalyzátoru a pomocí hydridu lithnohlinitého došlo k odstranění ketonové skupiny u 26. Pokračovalo se allylovým přesmykem na alkohol 27 (isostrychnin) za účasti roztoku bromovodíku v kyselině octové a hydrolýzy kyselinou sírovou. V posledním kroku k získání (−)-strychninu 28 sloučenina 27 reagovala s ethanolovým roztokem hydroxidu draselného, což vedlo k přesmyku na dvojné vazbě C12-13 a uzavření kruhu konjugovanou adicí hydroxylového aniontu.
Magnusova syntéza
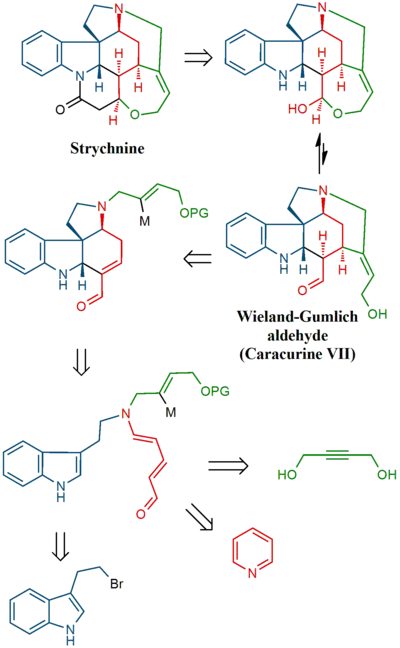
Při Magnusově syntéze byl nejprve připraven jeden z produktů rozkladu strychninu, který lze získat několika kroky z jiného produktu rozkladu, nazývaného Wielandův-Gumlichův aldehyd. V závěrečné části byl z této sloučeniny vytvořen strychnin.
Overmanova syntéza
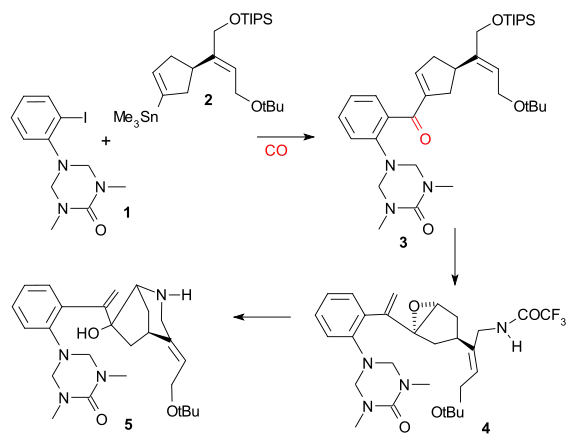
Larry E. Overman použil v roce 1993 jako výchozí látku chirální derivát cyklopentenu získaný enzymatickou hydrolýzou cis-1,4-diacetoxycyklopent-2-enu. V několika krocích jej přeměnil na trialkylstannan 2, který poté reagoval s aryljodidem 1 ve Stilleově reakci za přítomnosti oxidu uhelnatého, katalyzátory byly tris(dibenzylidenaceton)dipalladium a trifenylarsan. Sloučenina 3 byla přeměněna na epoxid reakcí s terc-butylhydroperoxidem, potom se karbonylová skupina změnila na alkenovou s využitím Wittigovy reakce Ph3P=CH2 a následně proběhla hydrolýza triisopropylsilylu (TIPS) tetra-n-butylamoniumfluoridem a její náhrada trifluoracetamidem (NH2COCF3) s využitím hydridu sodného (NaH) (4). Cyklizací pomocí NaH následně vedla k otevření epoxidového kruhu a trifluoracetylová skupina byla odstraněna hydroxidem draselným za vzniku azabicyklooktanu 5.
Důležitým krokem byla aza-Copeova-Mannichova reakce spuštěná aminkarbonylovou kondenzací za použití formaldehydu, kde se kvantitativně vytvořila sloučenina 6:
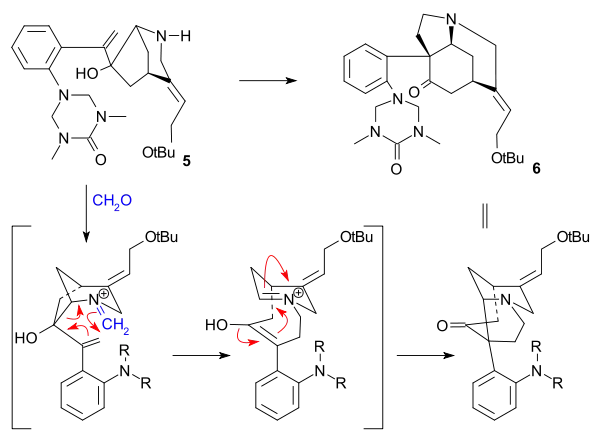
V závěrečné části byl získán strychnin skrz Wielandův-Gumlichův aldehyd (10):
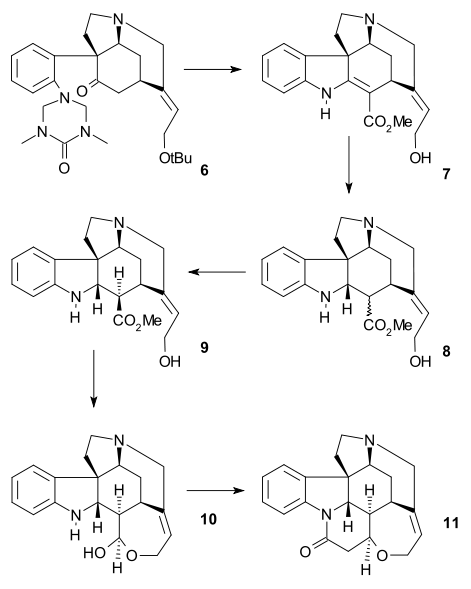
Meziprodukt 6 podstoupil acylaci methylkyanoformiátem a poté mohly být odvázány dvě chránicí skupiny pomocí HCl a MeOH (7). Dvojná vazba C8-C13 prošla redukcí zinkem (v MeOH za přítomnosti H+) na nasycený ester 8. Epimerizací na C13, provedenou methoxidem sodným v MeOH, vznikl beta-ester 9, jenž byl redukován diisobutylaluminiumhydridem na Wielandův-Gumlichův aldehyd 10. Přeměna tohoto aldehydu kyselinou malonovou na (−)-strychnin 11 již byla známa.
Kuehneova syntéza
Keuhneova syntéza z roku 1993 má za produkt racemický strychnin. Na začátku spolu reagují tryptamin 1 a 4,4-dimethoxy-akrolein 2 za přítomnosti fluoridu boritého na acetal 3 v amino-karbonylové kondenzaci, po níž následuje sigmatropní přesmyk.
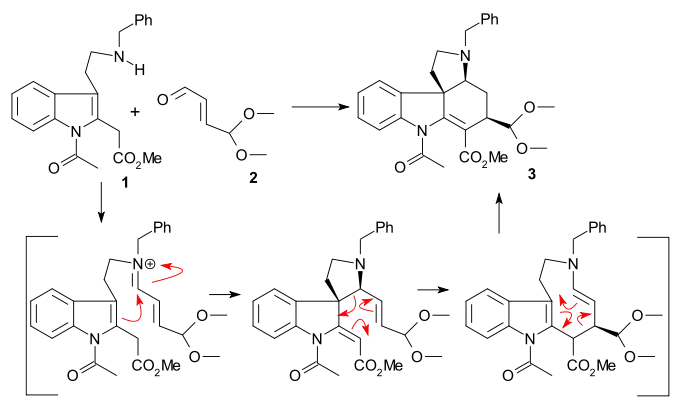
Hydrolýzou roztokem kyseliny chloristé vznikne aldehyd 4. Johnsonovou–Coreyovou–Čajkovského reakcí s trimethylsulfoniumjodidem a n-butyllithiem se aldehyd změnil na epoxid, jenž následně reagoval s terciárním aminem na kvartérní amonnou sůl 5 (vytvářely se i jiné produkty cyklizace). Redukcí vodíkem za katalýzy palladiem na uhlíku byla odstraněna benzylová skupina a vznikl alkohol 6, z něhož se další redukcí kyanoborohydridem sodným a acylací acetanhydridem v pyridinu vytvořil meziprodukt 7 jako směs epimerů (na C17). Uzavření kruhu III za tvorby 8 bylo provedeno aldolovou reakcí s bis(trimethylsilyl)amidem lithným (použit byl pouze epimer se správnou konfigurací). Poté následovala další redukce tetrahydridoboritanem sodným a acylace na epimerní diacetát 9.
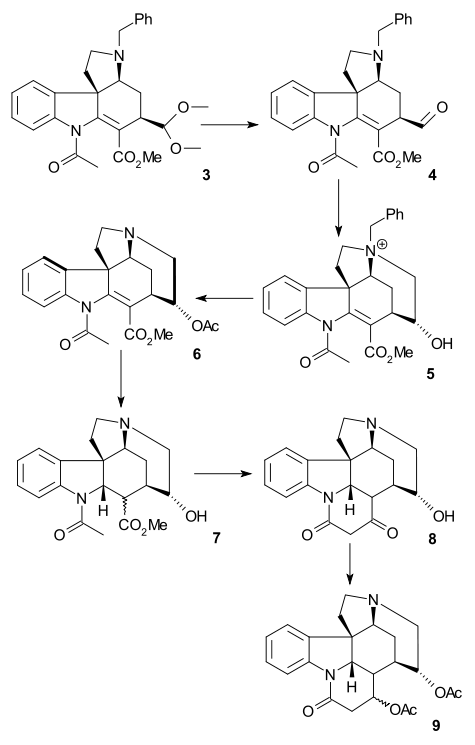
Eliminační reakcí řízenou diazabicykloundecenem (DBU) se vytvořil nenasycený alkohol 10, jenž Swernovou oxidací vytvořil nestabilní aminoketon 11. V závěrečné části proběhla Hornerova–Wadsworthova–Emmonsova reakce s methyl-2-(diethylfosfono)acetátem na akrylátový ester 12, který vznikl jako směs cis a trans izomerů, které byly na požadovaný trans izomer převedeny fotochemickým přesmykem, pak proběhla redukce esterové skupiny diisobutylaluminiumhydridem (DIBAL) a fluoridem boritým na isostrychnin 13, z nějž racemický strychnin 14 zásaditě katalyzovaným uzavřením kruhu stejně jako ve Woodwardově syntéze.
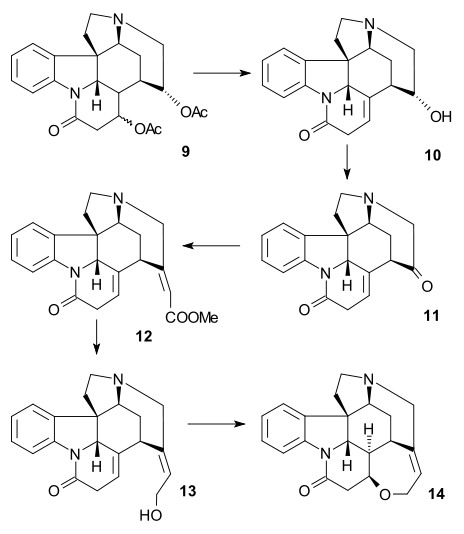
V roce 1998 získal Keuhne chirální (−)-strychnin z chirálního tryptofanu.
Rawalova syntéza
V Rawalově syntéze (racemického) strychninu zreagovaly amin 1 a enon 2 aminokarbonylovou kondenzací, následnou reakcí s methylchlorformiátem vznikl trien 3, jenž poté vstoupil do Dielsovy–Alderovy reakce (v benzenu při 185 °C) vytvářející hexen 4. Trojice esterových skupin byla hydrolyzována jodtrimethylsilanem, čímž vznikl po přidání methanolu a sedmi krocích (zahrnujících Dieckmannovu kondenzaci) pentacyklický laktam 5. Následně byl aminovou alkylací připojen čtyřuhlíkatý řetězec 6 a Heckovou reakcí sloučeniny 7 vznikl po odštěpení terc-butyldimethylsilylové (TBS) chránicí skupiny vytvořen isostrychnin 8.
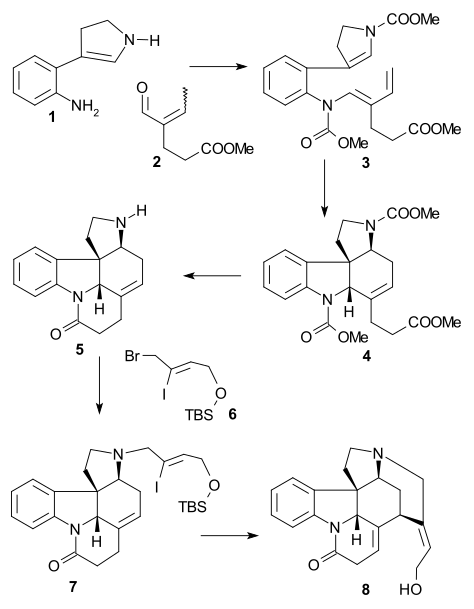
Celková výtěžnost dosáhla 10 % a byla tak nejvyšší, jaké se do té doby podařilo dosáhnout.[40]
Boschova synntéza
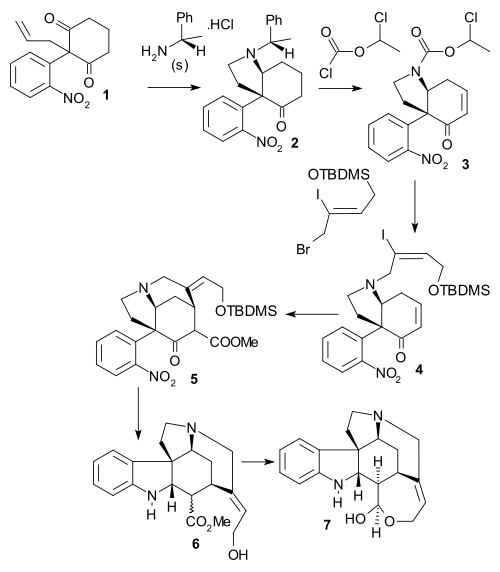
V Boschově syntéze chirálního strychninu (z roku 1999) byla nejprve alkenová skupina dionu 1 přeměněna ozonolýzou na aldehyd a po dvojité redukční aminaci (S)-1-fenylethylaminem vznikl chirální amin 2. Fenylethylový substituent byl odstraněn pomocí ClCO2CHClCH3 a Griecovou eliminací s trimethylsilyl jodidem (TMSI) a bis(trimethylsilyl)aminem (HMDS) byla zavedena enonová skupina. Dalšími reakcemi s fenylselenylchloridem (PhSeCl), ozonem a diisopropylaminem vznikl karbamát 3. Z aminu se odstranila chránicí skupina pomocí methanolu a poté proběhla alkylace (Z)-BrCH2CICH=CH2OTBDMS na terciární amin 4. Následovala Heckova reakce a po ní methoxykarbonylace bis(trimethylsilyl)amidem lithným a NCCO2Me na tricyklickou sloučeninou 5. Reakcí s práškovým zinkem v 10% kyselině sírové se odštěpila chránicí TBDMS skupina, redukcí vznikla nitrosloučenina a redukční aminokarbonylovou cyklizací vznikl tetracyklický meziprodukt 6 jako směs epimerů. V poslední části zreagoval Wielandův-Gumlichův aldehyd 7 s hydridem sodným v methanolu na správný epimer, který byl redukován na methylesterové skupině diisobutylaluminiumhydridem.
Vollhardtova syntéza
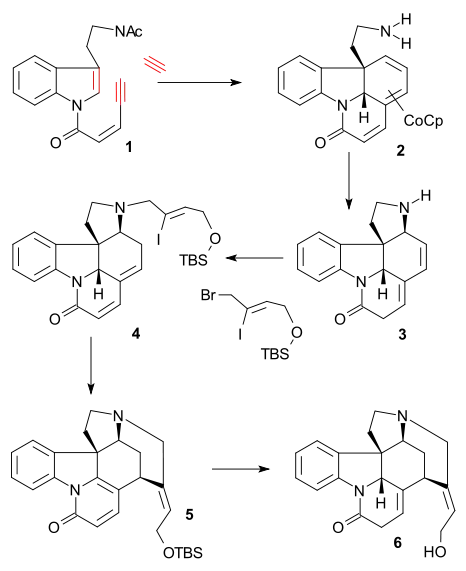
Důležitou součástí Vollhardtovy syntézy (racemického) strychninu je trimerizace derivátu tryptaminu 1 with acetylenem a CpCo(C2H4)2 v tetrahydrofuranu při 0 °C za vzniku tricyklické sloučeniny 2 a po odstranění aminové skupiny hydroxidem draselným ve směsi methanolu a vody. Následně došlo působením dusičnanu železitého k [1,8]-konjugované adici a tvorbě tetracyklické sloučeniny 3, aminovou alkylací (Z)-1-brom-4-[(terc-butyldimethylsilyl)oxy]-2-jodbut-2-enem (jako u Rawalovy syntézy) a uhličitanem lithným a následnou izomerizací dienu s využitím isopropoxidu draselného a isopropylalkoholu vznikl enone 4. Heckovou reakcí za přítomnosti octanu palladnatého a trifenylfosfinu se po následné aromatizaci vytvořil pyridon 5, z něhož se redukcí hydridem litnohlinitým po odstranění chránicí TBS skupiny vytvořil isostrychnin 6.
Moriova syntéza
Moriova syntéza, chirálního, (-)-strychninu, popsaná roku 2003, byla první syntézou obsahující asymetrický krok. Její součástí je také velký počet reakcí katalyzovaných palladiem.
Na začátku reaguje N-tosylamin 1 s allylkarbonátem 2 v Cudžiově–Trostově reakci za přítomnosti Pd2(dba)3 a asymetrického ligandu (S-BINAPO) na chirální sekundární amin 3. Desilylací TBDMS skupiny pomocí HCl za vzniku alkoholu, následně přeměněného na nitril 4 působením kyanidu sodného a bromidu fosforitého. Heckovou reakcí za přítomnosti octanu palladnatého a dimetylfenylfosfinu (Me2PPh) a debromací uhličitanem stříbrným se utvoří tricyklická sloučenina 5. Redukcí nitrilu na amin tetrahydridohlinitanem lithným a navázáním terc-butoxykarbonylové skupiny di-terc-butyldikarbonátem (Boc2O) 6 byla následována další allylovou oxidací, za přítomnosti octanu palladnatého, kyseliny octové, benzochinonu a oxidu manganičitého na tetracyklus 7. Hydroboračně-oxidační reakcí s využitím 9-borabicyklo[3.3.1]nonanu (9-BBN) a peroxidu vodíku vznikl alkohol 8, který byl Sewrnovou oxidací přeměněn na keton 9. Reakcí s diisopropylamidem lithným (LDA) a N-fenyltrifluormethansulfonimidem (PhNTf2) se vytvořil triflát enolu 10, z něho byla octanem palladnatým a trifenylfosfinem odštěpena triflátová skupina a vytvořil se alken 11.
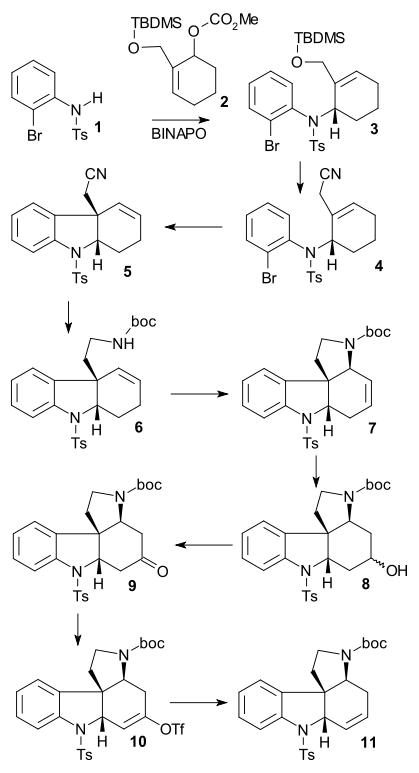
Detosylací 11 naftalenidem sodným a amidací 3-bromakryloylchloridem vznikl amid 12, poté se další Heckovou reakcí vytvořil pentacyklus 13. Izomerizací dvojné vazby sodíkem a isopropylalkoholem, odštěpením terc-butyloxykarbonylové (Boc) chránicí skupinykyselinou trifluormethansulfonovou a aminovou alkylací (Z)-BrCH2CICH=CH2OTBDMS se vytvořila sloučenina 14 (stejná jako jeden z Vollhardtových meziproduktů). Nakonec Heckovou reakcí (15) a odstraněním chránicí skupiny (TBDMS) vznikl (−)-isostrychnin 16.
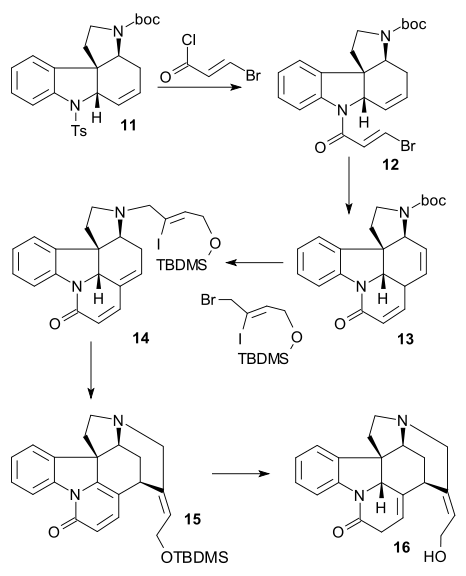
Šibasakiova syntéza
Šibasakiova syntéza (-)-chirálního strychninu byla druhou popsanou metodou využívající asymetrický krok. Na začátku reagoval cyklohexenon 1 s dimethylmalonátem v asymetrické Michaelově reakci za přítomnosti AlLibis(binaftoxidu) 2, přičemž vzniká chirální diester 3. Jeho ketonová skupina byla ochráněna jako acetal 2-ethyl-2-methyl-1,3-dioxolanem a kyselinou trifluormethansulfonovou a pomocí chloridu lithného v dimethylsulfoxidu při 140 °C byla odstraněna karboxylová skupina monoesteru 4. Poté byl přidán C2 fragment jako Weinrebův amid 5, čímž se vytvořil po přidáni diisopropylamidu lithného PMB ether 6. Keton byl poté kyanoborohydridem sodným a chloridem titaničitým redukován na alkohol, následně se odštěpila voda působením N,N'-dicyklohexylkarbodiimidu a chloridu měďného za vzniku alkenu 7. Po redukci esteru na alkohol diisobutylaluminiumhydridem a navázání triisopropylsilylové chránicí skupiny s využitím triisopropylsilyl-trifluoromethansulfonátu v triethylaminu, následovalo odstranění acetalu kyselinou kanforsulfonovou a tvorba ketonu 8. Poté se vytvořil Saegusovou–Itoovou oxidací enon 9. Jeho převedení na alkohol 10 zajistila Mukaijamova aldolová adice pomocí formaldehydu po níž proběhla, jodace jodem a 4-dimethylaminopyridinem. Její produkt 11 vstoupil do Stilleovy reakce katalyzované tris(dibenzylidenaceton)dipalladiem, trifenylarsinem a jodidem měďným, která vedla ke vzniku derivátu nitrobenzenu 12. Pak vznikl alkohol 13 navázáním SEM chránicí skupiny (pomocí SEMCl a i-Pr2NEt) a odštěpením triisopropylsilylu kyselinou fluorovodíkovou.
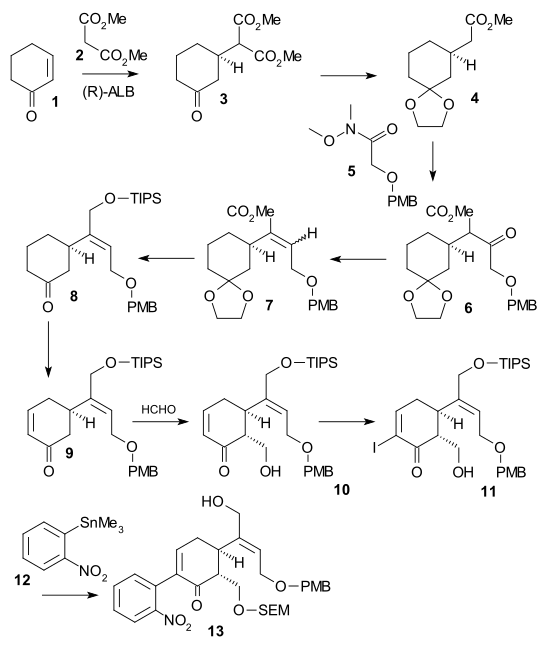
V druhé části se alkohol 13 změnil na triflát reakcí s anhydridem kyseliny trifluormethansulfonové za přítomnosti N,N-diisopropylethylaminu, následně byly přidány 2,2-bis(ethylthio)ethylamin 14 a práškový zinek, což spustilo tandemovou reakci vedoucí k redukci nitroskupiny, po 1,4-adici thioaminu a amino-ketokondenzaci se vytvořil indol 16. Reakcí dimethyl(methylthio)sulfoniumtetrafluorboritanu (DMTSF) došlo k thioniovému ataku na C7 za tvorby 17, iminová skupina této sloučeniny byla zredukována kyanoborohydridem sodným s chloridem titaničitým, nově získaná aminoskupina podstoupila acylaci acetanhydridem v pyridinu, proběhlo odštěpení obou alkoholových skupin methoxidem sodným v methanolu a allylalkoholová skupina byla znovu ochráněna triisopropylsilylem. Tímto se umožnilo odstranění ethylthioskupiny (provedené s využitím chloridem nikelnatým a tetrahydridoboritanem sodným ve směsi ethanolu a methanolu) za vzniku 18. Alkohol se zoxidoval na aldehyd Parikhovou–Doeringovou oxidací a oddělením triisopropylsilylové skupiny vznikl poloacetal 19 nazývaný (+)-diabolin, což je acylovaný Wielandův-Gumlichův aldehyd.
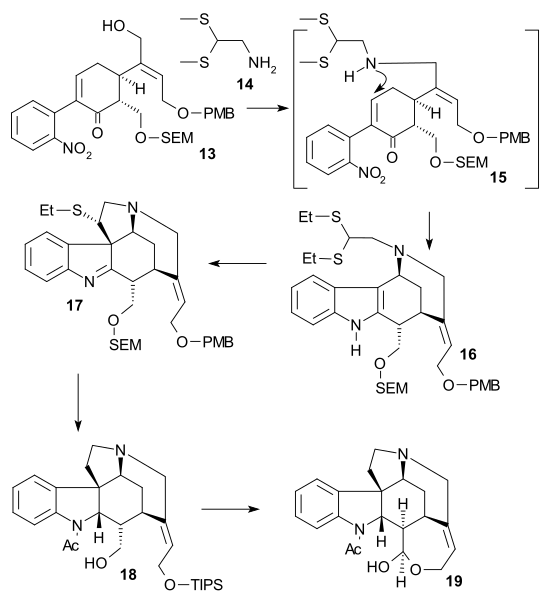
Liova syntéza
Liova syntéza racemického strychninu z roku 2002 vytvořila sloučeninu (číslo 5) již dříve vzniklou v Rawalově syntéze. Hlavní část obsahovala inverzní Dielsova–Alderova reakce of cyklofanu 1 vyvolaná jeho zahříváním s N,N-diethylanilinem (za odštěpení dusíku) následovaná redukcí dvojné vazby a tvorbou sloučeniny 2, jež se přeměnila na 3 tetrahydridoboritanem sodným s kyselinou trifluormethansulfonovou a odštěpení karbamátové chránicí skupiny dichpromanem pyridinia s křemelinou za tvorby 4.
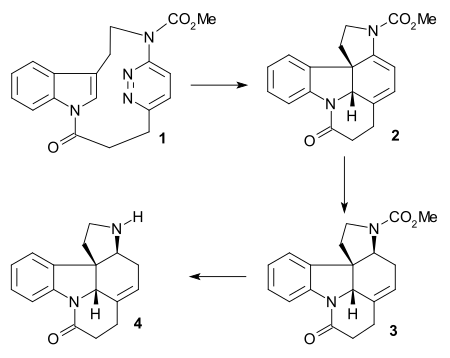
Fukujamova syntéza
Fukujamova syntéza (-)-strychninu, popsaná roku 2004, začínala u cyklického aminu 1. Chiralita byla do této látky zavedena enzymatickým rozlišením jednoho z prekurzorů. Poté vznikl acyloin 2 Rubottomovou oxidací a následnou hydrolýzou. Oxidačním štěpením pomocí octanu olovičitého se utvořil aldehyd 3, který po oddělení nosylové skupiny s využitím thiofenolu a uhličitanu cesného vyvolal aminkarbonylovou kondenzaci s iminiovým iontem 4, poté reakce pokračovala cyklizací na diester 5, jenž moohl být známými postupy přeměněn na Wielandův-Gumlichův aldehyd.
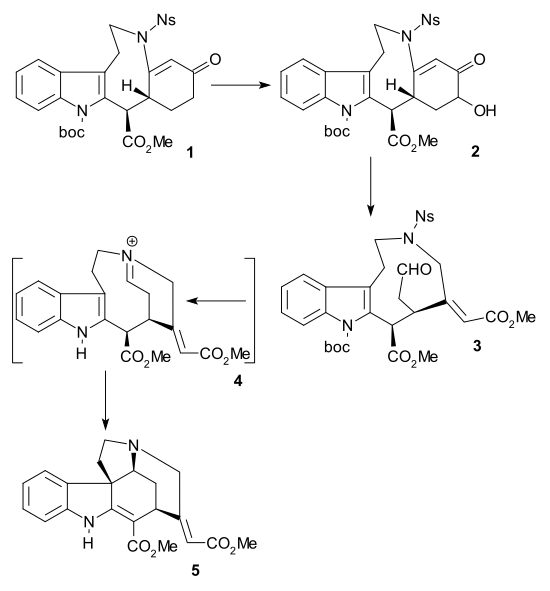
Reissigova syntéza
Reissigova syntéza racemického strychninu, popsaná v roce 2010, také vede k tvorbě Rawalova pentacyklu (viz amin 5 u Rawalovy syntézy). Indol 1 byl přeměněn na tetracyklus 2 (společně s ním vznikal vedlejší produkt) jedinou kaskádové reakci za přítomnosti jodidu samarnatého a hexamethylfosforamidu.[41] Redukcí vodíkem za katalýzy Raneyovým niklem vznikl amin 3 a jednonádobovou posloupností reakcí, nejprve s methylchlorformiátem, 4-dimethylaminopyridinem a triethylaminem, poté s methansulfonylchloridem, 4-dimethylaminopyridinem a triethylaminem a nakonec s 1,8-diazabicykloundec-7-enem vznikl Rawal§ův prekurzor 4 s atomy vodíku v potřebné anti konfiguraci.

Původně byl meziprodukt 2 nejprve redukován na imin 5 a následně převeden na karbamát 6 a dehydratován Burgessovým činidlem na dien 7, z něhož redukcí kyanoborohydridem sodným vznikla sloučenina 8 (sodium cyanoborohydride). Atomy vodíku u 8 mají nežádoucí cis-konfiguraci.
Vanderwalova syntéza
V roce 2011 získala skupina, kterou vedl Christopher D. Vanderwal, strychnin postupem s nejdelší lineární sekvencí tvořenou šesti kroky.[32] Její součástí byla Zinckeova reakce následovaná aniontovou bicyklizací a tandemem Brookova přesmyku s konjugovanou adicí.
Reference
V tomto článku byl použit překlad textu z článku Strychnine total synthesis na anglické Wikipedii.
- X-RAY; MESSERSCHMIDT, M.; SCHEINS, S.; LUGER, P. Charge density of (−)-strychnine from 100 to 15 K, a comparison of four data sets. Acta Crystallographica B. 2005, s. 115–121. DOI 10.1107/S0108768104032781. PMID 15659864. (anglicky)
- Nicolaou, K. C.; Sorensen, E. J. (1996). Classics in Total Synthesis: Targets, Strategies, Methods. Wiley ISBN 978-3-527-29231-8
- K. C. Nicolaou, Dionisios Vourloumis, Nicolas Winssinger, Phil S. Baran The Art and Science of Total Synthesis at the Dawn of the Twenty-First Century Angewandte Chemie International Edition 2000; Volume 39, Issue 1, Pages: 44-122
- BONJOCH, Josep; SOLE, Daniel. Synthesis of Strychnine. Chemical Reviews. 2000, s. 3455–3482. DOI 10.1021/cr9902547. PMID 11777429. (anglicky)
- PROUDFOOT, John R. Reaction Schemes Visualized in Network Form: The Syntheses of Strychnine as an Example. Journal of Chemical Information and Modeling. 2013, s. 1035–1042. DOI 10.1021/ci300556b. PMID 23597302. (anglicky)
- PELLETIER; CAVENTOU. Note sur un nouvel alkalai. Annales de Chimie et de Physique. 1818, s. 323–324. (anglicky)
- PELLETIER; CAVENTOU. Mémoire sur un nouvel alcali vegetal (la strychnine) trouvé dans la feve de Saint-Ignace, la noix vomique, etc.. Annales de Chimie et de Physique. 1819, s. 142–176. (anglicky)
- ROBINSON, R. The constitution of strychnine. Experientia. 1946, s. 1946. DOI 10.1007/BF02154708. PMID 21012825. (anglicky)
- BRIGGS, L. H.; OPENSHAW, H. T.; ROBINSON, Robert. Strychnine and brucine. Part XLII. Constitution of the neo-series of bases and their oxidation products. Journal of the Chemical Society. 1946, s. 903. DOI 10.1039/JR9460000903. (anglicky)
- OPENSHAW, H. T.; ROBINSON, R. Constitution of Strychnine and the Biogenetic Relationship of Strychnine and Quinine. Nature. 1946, s. 438. DOI 10.1038/157438a0. PMID 21024272. Bibcode 1946Natur.157..438O. (anglicky)
- WOODWARD, R. B.; BREHM, Warren J.; NELSON, A. L. The Structure of Strychnine. Journal of the American Chemical Society. 1947, s. 2250. DOI 10.1021/ja01201a526. PMID 20262753. (anglicky)
- Bijvoet, Schoone and Bokhoven, Kon. Ned. Akad. Wet., 50, No 8, 51, No. 8, 52, No. 2 (1947–49)
- BOKHOVEN, C.; SCHOONE, J. C.; BIJVOET, J. M. The Fourier synthesis of the crystal structure of strychnine sulphate pentahydrate. Acta Crystallographica. 1951, s. 275–280. Dostupné online. DOI 10.1107/S0365110X51000891. (anglicky)
- ROBERTSON, J. H.; BEEVERS, C. A. Crystal Structure of Strychnine Hydrobromide. Nature. 1950, s. 690–691. DOI 10.1038/165690a0. PMID 15416785. Bibcode 1950Natur.165..690R. (anglicky)
- ROBERTSON, J. H.; BEEVERS, C. A. The crystal structure of strychnine hydrogen bromide. Acta Crystallographica. 1951, s. 270–275. DOI 10.1107/S0365110X5100088X. (anglicky)
- WOODWARD, R. B.; CAVA, Michael P.; OLLIS, W. D.; HUNGER, A.; DAENIKER, H. U.; SCHENKER, K. The Total Synthesis of Strychnine. Journal of the American Chemical Society. 1954, s. 4749–4751. DOI 10.1021/ja01647a088. (anglicky)
- WOODWARD, R. B.; CAVA, M. P.; OLLIS, W. D.; HUNGER, A.; DAENIKER, H. U.; SCHENKER, K. The total synthesis of strychnine. Tetrahedron. 1963, s. 247–288. DOI 10.1016/s0040-4020(01)98529-1. (anglicky)
- MAGNUS, Philip; GILES, Melvyn; BONNERT, Roger; KIM, Chung S.; MCQUIRE, Leslie; MERRITT, Andrew; VICKER, Nigel. Synthesis of strychnine via the Wieland-Gumlich aldehyde. Journal of the American Chemical Society. 1992, s. 4403–4405. DOI 10.1021/ja00037a058. (anglicky)
- KNIGHT, Steven D.; OVERMAN, Larry E.; PAIRAUDEAU, Garry. Synthesis applications of cationic aza-Cope rearrangements. 26. Enantioselective total synthesis of (−)-strychnine. Journal of the American Chemical Society. 1993, s. 9293–9294. DOI 10.1021/ja00073a057. (anglicky)
- KUEHNE, Martin E.; XU, Feng. Total synthesis of strychnan and aspidospermatan alkaloids. 3. The total synthesis of (+-)-strychnine. The Journal of Organic Chemistry. 1993, s. 7490–7497. DOI 10.1021/jo00078a030. (anglicky)
- KUEHNE, Martin E.; XU, Feng. Syntheses of Strychnan- and Aspidospermatan-Type Alkaloids. 10. An Enantioselective Synthesis of (−)-Strychnine through the Wieland−Gumlich Aldehyde. The Journal of Organic Chemistry. 1998, s. 9427–9433. DOI 10.1021/jo9813989. (anglicky)
- RAWAL, Viresh H.; IWASA, Seiji. A Short, Stereocontrolled Synthesis of Strychnine. The Journal of Organic Chemistry. 1994, s. 2685–2686. DOI 10.1021/jo00089a008. (anglicky)
- Total Synthesis of (−)-Strychnine via the Wieland-Gumlich Aldehyde Angewandte Chemie International Edition Volume 38, Issue 3, 1999, Pages: 395-397 Daniel Solé, Josep Bonjoch, Silvina García-Rubio, Emma Peidró, Joan Bosch
- SOLÉ, Daniel; BONJOCH, Josep; GARCÍA-RUBIO, Silvina; PEIDRÓ, Emma; BOSCH, Joan. Enantioselective Total Synthesis of Wieland-Gumlich Aldehyde and (−)-Strychnine. Chemistry: A European Journal. 2000, s. 655–665. DOI 10.1002/(SICI)1521-3765(20000218)6:4<655::AID-CHEM655>3.0.CO;2-6. (anglicky)
- EICHBERG, Michael J.; DORTA, Rosa L.; LAMOTTKE, Kai; VOLLHARDT, K. Peter C. The Formal Total Synthesis of (±)-Strychnine via a Cobalt-Mediated [2 + 2 + 2]Cycloaddition. Organic Letters. 2000, s. 2479–2481. DOI 10.1021/ol006131m. PMID 10956526. (anglicky)
- EICHBERG, Michael J.; DORTA, Rosa L.; GROTJAHN, Douglas B.; LAMOTTKE, Kai; SCHMIDT, Martin; VOLLHARDT, K. Peter C. Approaches to the Synthesis of (±)-Strychnine via the Cobalt-Mediated [2 + 2 + 2] Cycloaddition: Rapid Assembly of a Classic Framework. Journal of the American Chemical Society. 2001, s. 9324–9337. DOI 10.1021/ja016333t. PMID 11562215. (anglicky)
- NAKANISHI, Masato; MORI, Miwako. Total Synthesis of (−)-Strychnine. Angewandte Chemie International Edition. 2002, s. 1934–1936. DOI 10.1002/1521-3773(20020603)41:11<1934::AID-ANIE1934>3.0.CO;2-F. PMID 19750638. (anglicky)
- MORI, Miwako; NAKANISHI, Masato; KAJISHIMA, Daisuke; SATO, Yoshihiro. A Novel and General Synthetic Pathway to Strychnos Indole Alkaloids: Total Syntheses of (−)-Tubifoline, (−)-Dehydrotubifoline, and (−)-Strychnine Using Palladium-Catalyzed Asymmetric Allylic Substitution. Journal of the American Chemical Society. 2003, s. 9801–9807. DOI 10.1021/ja029382u. PMID 12904045. (anglicky)
- OHSHIMA, Takashi; XU, Youjun; TAKITA, Ryo; SHIMIZU, Satoshi; ZHONG, Dafang; SHIBASAKI, Masakatsu. Enantioselective Total Synthesis of (−)-Strychnine Using the Catalytic Asymmetric Michael Reaction and Tandem Cyclization. Journal of the American Chemical Society. 2002, s. 14546–14547. DOI 10.1021/ja028457r. PMID 12465959. (anglicky)
- BODWELL, Graham J.; LI, Jiang. A Concise Formal Total Synthesis of (±)-Strychnine by Using a Transannular Inverse-Electron-Demand Diels–Alder Reaction of a [3](1,3)Indolo[3](3,6)pyridazinophane. Angewandte Chemie International Edition. 2002, s. 3261–3262. DOI 10.1002/1521-3773(20020902)41:17<3261::AID-ANIE3261>3.0.CO;2-K. (anglicky)
- KABURAGI, Y.; TOKUYAMA, H.; FUKUYAMA, T. Total synthesis of (−)-strychnine. Journal of the American Chemical Society. 2004, s. 10246–10247. DOI 10.1021/ja046407b. PMID 15315428. (anglicky)
- MARTIN, David B. C.; VANDERWAL, Christopher D. A synthesis of strychnine by a longest linear sequence of six steps. Chemical Science. 2011, s. 649. DOI 10.1039/C1SC00009H. (anglicky)
- JONES, Spencer B.; SIMMONS, Bryon; MASTRACCHIO, Anthony; MACMILLAN, David W. C. Collective synthesis of natural products by means of organocascade catalysis. Nature. 2011, s. 183–188. DOI 10.1038/nature10232. PMID 21753848. (anglicky)
- KNIGHT, Steven D.; OVERMAN, Larry E.; PAIRAUDEAU, Garry. Asymmetric Total Syntheses of (−)- and (+)-Strychnine and the Wieland-Gumlich Aldehyde. Journal of the American Chemical Society. 1995, s. 5776–5788. DOI 10.1021/ja00126a017. (anglicky)
- ZHANG, Hongjun; BOONSOMBAT, Jutatip; PADWA, Albert. Total Synthesis of (±)-Strychnine via a [4 + 2]-Cycloaddition/Rearrangement Cascade. Organic Letters. 2007, s. 279–282. DOI 10.1021/ol062728b. PMID 17217284. (anglicky)
- SIRASANI, Gopal; PAUL, Tapas; WILLIAM DOUGHERTY; KASSEL, Scott; ANDRADE, Rodrigo B. Concise Total Syntheses of (±)-Strychnine and (±)-Akuammicine. The Journal of Organic Chemistry. 2010, s. 3529–3532. DOI 10.1021/jo100516g. PMID 20408591. (anglicky)
- BEEMELMANNS, C.; REISSIG, H.-U. A Short Formal Total Synthesis of Strychnine with a Samarium Diiodide Induced Cascade Reaction as the Key Step. Angewandte Chemie International Edition. 2010, s. 8021–8025. DOI 10.1002/anie.201003320. PMID 20848626. (anglicky)
- R. Robinson "Molecular structure of Strychnine, Brucine and Vomicine Progress in Organic Chemistry, 1952; 1 ,2
- WOODWARD, R. B. Biogenesis of the Strychnos Alkaloids. Nature. 1948, s. 155–156. DOI 10.1038/162155a0. PMID 18871488. Bibcode 1948Natur.162..155W. (anglicky)
- CANNON, J. S.; OVERMAN, L. E. Is There No End to the Total Syntheses of Strychnine? Lessons Learned in Strategy and Tactics in Total Synthesis. Angewandte Chemie International Edition. 2012, s. 4288–4311. DOI 10.1002/anie.201107385. PMID 22431197. (anglicky)
- SZOSTAK, M.; PROCTER, D. J. Concise Syntheses of Strychnine and Englerin A: the Power of Reductive Cyclizations Triggered by Samarium Iodide. Angewandte Chemie International Edition. 2011, s. 7737–7739. DOI 10.1002/anie.201103128. PMID 21780264. (anglicky)