Aldolová reakce
Aldolová reakce je organická reakce a jeden ze způsobů tvorby vazeb mezi uhlíkovými atomy;[1][2][3] objevili ji nezávisle na sobě roku 1869 ruský chemik Alexandr Borodin a v roce 1872 francouzský chemik Charles Adolphe Wurtz,[4][5][6] Při této reakci vzájemně reagují dvě karbonylové sloučeniny (v původních experimentech to byly dva aldehydy) za vzniku β hydroxykarbonylového produktu, takto vzniklé látky se nazývají aldoly. Aldolové skupiny jsou součástí molekul mnoha přírodních i syntetických sloučenin.[7][8][9] Aldolová reakce se využívá například při výrobě pentaerythritolu[10] a léčiva na srdeční onemocnění atorvastatinu.[11][12]
Při aldolové reakci se spojují dvě poměrně jednoduché molekuly za vzniku jedné složitější. Zvýšení složitosti molekuly je způsobeno tvorbou dvou nových stereocenter na α- a β uhlíku (označených hvězdičkami na níže zobrazeném obrázku) vzniklého aldolového aduktu. Moderní metodologie umožňuje nejen dosáhnout při aldolové reakci vysoké výtěžnosti, ale též ovlivňovat relativní a absolutní konfiguraci těchto stereocenter.[13] Možnost selektivně vytvořit určitý stereoizomer je důležitá, protože různé stereoizomery mohou mít značně odlišné chemické a biologické vlastnosti.
Jako příklad přírodních látek obsahujících stereogenní aldolové skupiny lze uvést polyketidy; ty se v přírodě syntetizují pomocí enzymů provádějících Claisenovy kondenzace. 1,3-dikarbonylové produkty těchto reakcí mohou být různě pozměňovány za vzniku řady dalších sloučenin; pokud taková změna proběhne na karbonylu, tak se často vytvoří aldolová skupina. Mnohé takto vytvořené látky mají důležité biologické vlastnosti, patří k nim například imunosupresivum FK506 a antimykotikum amfotericin B. I když byla syntéza některých takových látek dříve považována za téměř nemožnou, tak je nyní lze v mnoha případech účinně syntetizovat.[14]
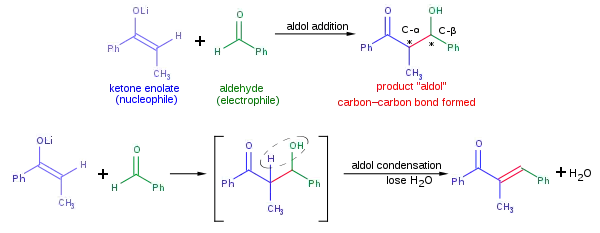
Běžná adiční reakce, znázorněná na obrázku výše, spočívá v nukleofilní adici ketonenolátu na aldehyd. Vzniklý aldol může podléhat dehydratační reakci za vzniku α,β nenasycené karbonylové sloučeniny; tato reakce se nazývá aldolová kondenzace. Při aldolové reakci lze použít mnoho různých nukleofilů, mimo jiné enoly, enoláty a enolethery aldehydů či ketonů, a řadu dalších karbonylových sloučenin; elektrofilem je zde obvykle aldehyd nebo keton. Pokud jsou nukleofil a elektrofil rozdílné látky, reakce se nazývá zkřížená aldolová reakce, pokud jde o stejné látky, tak je reakce aldolovou dimerizací.
Mechanismy
Aldolová reakce může proběhnout dvěma zcela odlišnými mechanismy; při prvním jsou karbonylové sloučeniny převedeny na enoly či enolethery, ty mají nukleofilní α uhlík a dále reagují s protonovanými karbonylovými sloučeninami, například protonovanými aldehydy – v tomto případě jde o enolový mechanismus. Karbonylové sloučeniny mohou být také deprotonovány na enoláty, které jsou mnohem nukleofilnější než enoly a enolethery, a mohou přímo reagovat s elektrofilem (což je zde obvykle aldehyd, jelikož aldehydy jsou výrazně reaktivnější než ketony) – pak jde o enolátový mechanismus.
Za určitých podmínek může dojít ke kondenzaci, čemuž se však dá zabránit použitím vhodných činidel (například diisopropylamidu lithného, což je silná zásada, v THF) a provedením reakce při nízké teplotě. I když aldolová reakce probíhá téměř úplně a je nevratná, tak jsou izolované aldoly náchylné k zásadami indukovaným zpětným reakcím.[15]

Enolový mechanismus
Při kyselé katalýze je úvodním krokem reakčního mechanismu tautomerizace karbonylové sloučeniny na enol. Kyselina slouží nejen jako katalyzátor tautomerizace, ale i jako aktivátor karbonylové skupiny další molekuly prostřednictvím protonace, díky čemuž je tato skupina značně elektrofilní. Enol má na α uhlíku nukleofilní centrum, což mu umožňuje reagovat s karbonylovou sloučeninou za vzniku aldolu po následné deprotonaci; ten se poté často dehydratuje za vzniku nenasycené karbonylové sloučeniny.
Enolátový mechanismus
Pokud je katalyzátorem aldolové reakce zásada jako například hydroxid nebo alkoxid, pak tato reakce začíná nukleofilním atakem rezonančně stabilizovaného enolátu na karbonylovou skupinu další molekuly, přičemž se vytváří alkoxidová sůl aldolu. Následně vznikne samotný aldol, jenž opět může být dehydratován na nenasycenou karbonylovou sloučeninu.
I když někdy stačí pouze katalytická množství zásady, tak je častěji třeba použít stechiometrické množství silné zásady, jako je například diisopropylamid lithný nebo hexamethyldisilazid sodný. V tomto případě je tvorba enolátu nevratná a aldol nevznikne, dokud není alkoxidová sůl protonována v odděleném reakčním kroku.
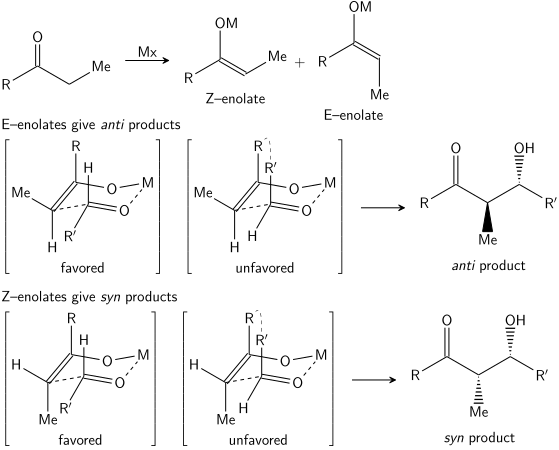
Zimmermanův–Traxlerův model
V roce 1957 Howard Zimmerman a M. D. Traxler oznámili, že při některých aldolových reakcích vznikají „šestičlenné přechodné stavy s židličkovou konformací“,[16] tento model byl později nazván Zimmermanův–Traxlerův model. Z E-enolátů vznikají anti izomery, zatímco ze Z-enolátů se tvoří syn produkty; selektivita je způsobena přednostním umísťováním substituentů do ekvatoriálních poloh vzhledem k šestičlennému cyklu a podporuje ji také tendence molekul vyhnout se syn-pentanovým inteerakcím.[17]
V praxi tomuto modelu odpovídají jen některé kovy, mimo jiné lithium, v některých případech se tak stereochemie produktů nedá předvídat.
Stereoselektivita
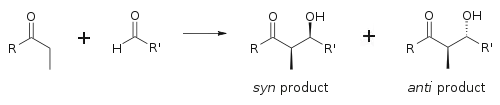
Aldolová reakce je obzvláště výhodná, jelikož při jedné reakci vznikají dvě stereogenní centra. K určení stereochmie na α- a β uhlíku se zde používá označování syn/anti.
Geometrie enolátů
Míra stereoindukce se u E- a Z enolátů příliš neliší. Každá z možných geometrií alkenu vede převážně k tvorbě jednoho ze dvou stereoizomerů výsledného produktu, z E alkenu vznikají anti- a ze Z alkenu syn produkty:[18]
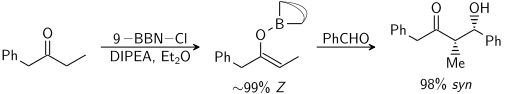
Vliv kationtů v enolátu
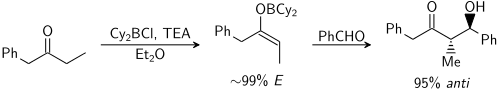
Kation v molekule enolátu může značně ovlivňovat míru stereoselektivity aldolové reakce; často se takto používá bor,[19][20] jelikož je u něj vazba výrazně kratší než u kovů jako jsou lithium, hořčík a hliník.

Například vazba B-C má délku kolem 145 pm a vazba B-O okolo 155 pm, zatímco vazba kov-kyslík má většinou délku 190 až 220 pm a vazba kov-uhlík 200 až 220 pm. Použití boru místo kovu „utáhne“ přechodnzý stav a zvýší stereoselektivitu reakce.[21] U výše zobrazené reakce je při použití lithného enolátu poměr syn:anti přibližně 80:20, zatímco při použití bibutylboritého enolátu má tento poměr hodnotu 97:3.
Evansova oxazolidinonová metoda
U moderních organosyntetických metod se vyžaduje příprava látek v enantiomerně čisté podobě. Jelikož aldolová reakce vytváří dvě stereocentra, tak mohou vzniknout až čtyři stereoizomery.
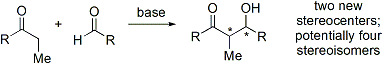
Bylo vyvinuto několik způsobů ovlivňování relativní (například syn/anti) i absolutní (například R/S) stereochemie.

Často používanou metodou je Evansova acylová oxazolidinonová metoda,[22][23] kterou vyvinul na přelomu 70. a 80. let 20. století David Evans se svými spolupracovníky. Postup spočívá v tvorbě chirálního enolátu s využitím chirálního pomocníka. Předem vytvořená chiralita v pomocné molekule se prostřednictvím diastereoselektivní aldolové reakce přenáší na aldolový adukt. Po odstranění chirálního pomocníka vzniká konečný produkt.

U této metody je chirálním pomocníkem oxazolidinon a vzniklá karbonylová sloučenina je imidem. Řada oxazolidinonů je dostupná v obou enantiomerních formách. Enantiomerně čisté oxazolidinony se vyrábějí ve dvou krocích z aminokyselin, obvykle edukcí za přítomnosti borohydridu, po níž následuje kondenzace či cyklizace vzniklého aminoalkoholu pomocí esteru kyseliny uhličité.
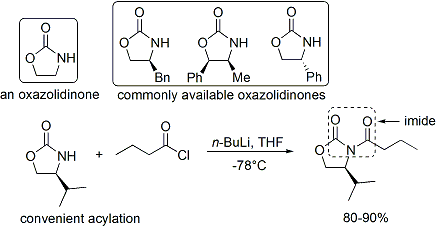
Často se provádí acylace oxazolidinonu. Z enoláty, z nichž vznikají syn produkty, lze připravit enolizací řízenou borem.
Evansovou metodou nelze účinně získat anti aldolové adukty. I přes nákladnost a možnost vytvářet pouze syn adukty se tato metoda díky své snadnosti a univerzálnosti velmi často.[25]
Při vytvoření imidu lze vytvořit syn- i anti produkty. Je možné provést syn selektivní[26] i anti selektivní reakci.[27]
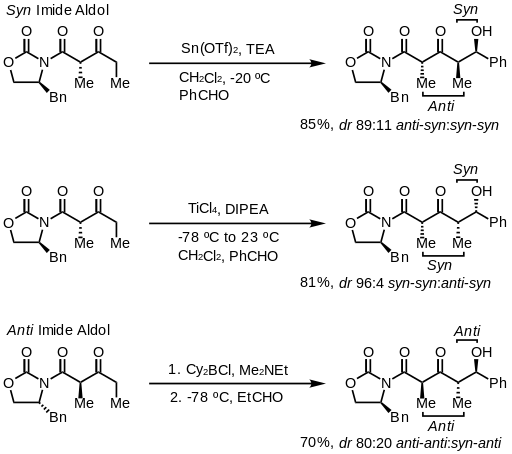
U syn selektivních reakcí vznikají při použití obou enolizačních metod Z produkty, jak se dá očekávat, stereochemii produktů ovšem ovlivňuje spíše methylové stereocentrum než chiralita oxazolidinonu.
Vnitromolekulární aldolová reakce

Vnitromolekulární aldolová reakce je vzájemná kondenzační reakce dvou aldehydových nebo ketonových skupin uvnitř jedné molekuly; při takové reakci vznikají α,β nenasycené ketony či aldehydy s pěti- nebo šestičlennými cykly. Jedná se o významný způsob tvorby vazeb mezi uhlíkovými atomy u organických sloučenin s cyklickými řetězci. Jako příklad lze uvést reakci hexan-2,5-dionu v zásaditém prostředí (sloučenina A na následujícím obrázku), kdy se tato látka cyklizuje při vnitromolekulůární aldolové reakci za vzniku 3-methylcyklopent-2-en-1-onu (sloučeniny B)
Mechanismus této reakce zahrnuje tvorbu enolátového meziproduktu a vnitromolekulární nukleofilní adici. Hydroxid nejprve způsobí odštěpení α vodíku na šestém uhlíku, čímž se vytvoří enolát. Následně v důsledku nukleofilního ataku enolátu na druhý ketonový uhlík vznikne vazba uhlík-uhlík mezi druhým a šestým uhlíkem. Nakonec je reakční směs zahřívána a odštěpením molekuly vody se vytvoří cyklický α,β nenasycený keton.
Vnitromolekulární aldolové reakce se často provádějí při totálních syntézách řady přírodních látek, nejčastěji u alkaloidů a steroidů. Příkladem může být níže zobrazená syntéza (+) wortmanninu:
Další varianty
Moderní metodologie umožňuje provést značné množství různých variant aldolové reakce, často s využitím katalytických množství chirálních ligandů. Pokud se při reakci použije malé množství enantiomerně čistých ligandů za účelem tvorby enantiomerně čistých produktů, tak se reakce často označuje jako „asymetrická“; lze použít mnoho různých katalyzátorů a pomocí nich provádět asymetrické aldolové reakce.
Acetátová aldolová reakce
Hlavním omezením výše popsaného využití chirálních pomocníků je nemožnost selektivní reakce u N-acetylimidů. Jednou z možností vyřešení tohoto problému je použití thioetheru.[25] V této reakci je nukleofilem enolát boru vzniklý z dibutylbortriflátu (nBu2BOTf), zásadou je N,N-diisopropylethylamin. Thioether se odstraní ve druhém kroku redukcí Raneyovým niklem.
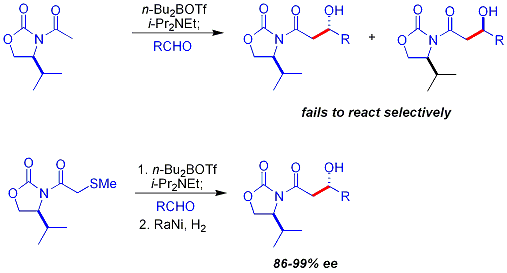
Mukaiyamova aldolová reakce
Mukaiyamova aldolová reakce[29] je nukleofilní adice silylenoletheru na aldehyd katalyzovaná Lewisovou kyselinou jako je například fluorid boritý nebo chlorid titaničitý.[30][31] Tato reakce neprobíhá podle Zimmermanova–Traxlerova modelu.
Byla popsána asymetrická metoda ukaiyamovy aldolové reakce za použití silylketenacetalu, která se vyznačuje vysokou enantioselektivitou a možností použít řadu různých substrátů.[32] Při tomto postupu se pracuje s nerozvětvenými alifatickými aldehydy, které jsou často pro asymetrické procesy příliš slabými elektrofily.

Organokatalytická aldolová reakce
Byla vyvinuta varianta aldolové reakce, při níž se jako katalyzátor používají chirální sekundární aminy. Tyto aminy vytváří reakcí s ketony enaminové meziprodukty, které následně mohou reagovat enantioselektivně.[33] s vhodnými aldehydovými elektrofily. Amin reaguje s karbonylovou sloučeninou za vzniku enaminu, který je nukleofilem, a z produktu se následně uvolní původní amin, který je tedy katalyzátorem. Často používaným aminem je zde prolin.
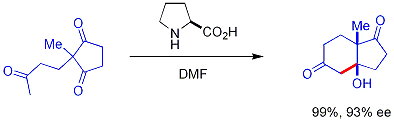
Výše zobrazená reakce se nazývá Hajosova-Parrishova reakce.[34][35][36] U této reakce je potřeba pouze katalytické množství prolinu (3 molární %). Nedochází zde k nechirálním vedlejším reakcím, jelikož jsou enaminové meziprodukty mnohem silnějšími nukleofily než ketoenoly, ze kterých vznikly. Tímto způsobem lze provádět enantioselektivní reakce bez použití přechodných kovů, které mohou být toxické nebo drahé.
Aldolové reakce katalyzované prolinem nevykazují nelineární jevy (enantioselektivita produktů přímo souvisí s enantiomerní čistotou katalyzátoru). Kombinací izotopového značkování a studií provedených pomocí metod výpočetní chemie bylo zjištěno, že mechanismus takovýchto reakcí je následující:[37]

Biologické aldolové reakce
Aldolovými reakcemi probíhajícími v živých organismech jsou například štěpení fruktóza-1,6-bisfosfátu na dihydroxyaceton a glyceraldehyd-3-fosfát během glykolýzy, enzymem katalyzujícím reakci je aldoláza A (fruktóza-1,6-bisfosfátaldoláza).
V glyoxylátovém cyklu, probíhajícím u rostlin a některých prokaryot, vzniká z isocitrátu působením isocitrátlyázy glyoxylát a sukcinát. Dochází zde k deprotonaci hydroxylu, po níž se isocitrát rozštěpí aldolovou reakcí, která je mechanisticky podobná výše uvedené eakci v glykolýze.
Reference
V tomto článku byl použit překlad textu z článku Aldol reaction na anglické Wikipedii.
- WADE, L. G. Organic Chemistry. 6th. vyd. Upper Saddle River, New Jersey: Prentice Hall, 2005. ISBN 0-13-236731-9. S. 1056–66. (anglicky)
- SMITH, M. B.; MARCH, J. Advanced Organic Chemistry. 5th. vyd. New York: Wiley Interscience, 2001. ISBN 0-471-58589-0. S. 1218–23. (anglicky)
- MAHRWALD, R. Modern Aldol Reactions, Volumes 1 and 2. Weinheim, Germany: Wiley-VCH Verlag GmbH & Co. KGaA, 2004. Dostupné online. ISBN 3-527-30714-1. S. 1218–23. (anglicky)
- WURTZ, C. A. Sur un aldéhyde-alcool. Bulletin de la Société Chimique de Paris. 1872, s. 436–442. Dostupné online. (French)
- WURTZ, C. A. Ueber einen Aldehyd-Alkohol. Journal für Praktische Chemie. 1872, s. 457–464. Dostupné online. DOI 10.1002/prac.18720050148. (German)
- WURTZ, C. A. Sur un aldéhyde-alcool. Comptes rendus de l'Académie des sciences. 1872, s. 1361. Dostupné online. (French)
- HEATHCOCK, C. H. Comprehensive Organic Synthesis. Redakce Trost, B. M.. [s.l.]: Elsevier Science, 1991. Dostupné online. ISBN 978-0-08-052349-1. DOI 10.1016/B978-0-08-052349-1.00027-5. Kapitola The Aldol Reaction: Acid and General Base Catalysis, s. 133–179. (anglicky)
- Mukaiyama T. The Directed Aldol Reaction. Org. React.. 1982, s. 203–331. ISBN 0471264180. DOI 10.1002/0471264180.or028.03. (anglicky)
- Paterson, I. New Asymmetric Aldol Methodology Using Boron Enolates. Chem. Ind.. 1988, s. 390–394. (anglicky)
- Mestres R. A green look at the aldol reaction. Green Chemistry. 2004, s. 583–603. DOI 10.1039/b409143b. (anglicky)
- M. Braun; R. DEVANT. (R) and (S)-2-acetoxy-1,1,2-triphenylethanol – effective synthetic equivalents of a chiral acetate enolate. Tetrahedron Letters. 1984, s. 5031–4. DOI 10.1016/S0040-4039(01)91110-4. (anglicky)
- Jie Jack Li. Contemporary Drug Synthesis. [s.l.]: Wiley-Interscience, 2004. ISBN 0-471-21480-9. S. 118–. (anglicky)
- Wulff W. D.; ANDERSSON B. A. Stereoselective aldol addition reactions of Fischer carbene complexes via electronic tuning of the metal center for enolate reactivity. Inorganica Chimica Acta. 1994, s. 215–231. DOI 10.1016/0020-1693(94)03874-0. (anglicky)
- Schetter, B.; MAHRWALD, R. Modern Aldol Methods for the Total Synthesis of Polyketides. Angew. Chem. Int. Ed.. 2006, s. 7506–7525. DOI 10.1002/anie.200602780. PMID 17103481. (anglicky)
- Guthrie, J.P.; COOPER, K.J.; COSSAR, J.; DAWSON, B.A.; TAYLOR, K.F. The retroaldol reaction of cinnamaldehyde. Canadian Journal of Chemistry. 1984, s. 1441–1445. DOI 10.1139/v84-243. (anglicky)
- Zimmerman, H. E.; TRAXLER, M. D. The Stereochemistry of the Ivanov and Reformatsky Reactions. I. Journal of the American Chemical Society. 1957, s. 1920–1923. DOI 10.1021/ja01565a041. (anglicky)
- HEATHCOCK, C. H.; BUSE, C. T.; KLESCHNICK, W. A.; PIRRUNG, M. C.; SOHN, J. E.; LAMPE, J. Acyclic stereoselection. 7. Stereoselective synthesis of 2-alkyl-3-hydroxy carbonyl compounds by aldol condensation. Journal of Organic Chemistry. 1980, s. 1066–1081. DOI 10.1021/jo01294a030. (anglicky)
- BROWN, H. C.; DHAR, R. K.; BAKSHI, R. K.; PANDIARAJAN, P. K.; SINGARAM, B. Major effect of the leaving group in dialkylboron chlorides and triflates in controlling the stereospecific conversion of ketones into either E- or Z-enol borinates. Journal of the American Chemical Society. 1989, s. 3441–3442. DOI 10.1021/ja00191a058. (anglicky)
- Cowden, C. J.; Paterson, I. Organic Reactions 1997, 51, 1.
- COWDEN, C. J.; PATERSON, I. Asymmetric Aldol Reactions Using Boron Enolates. [s.l.]: [s.n.], 2004. DOI 10.1002/0471264180.or051.01. (anglicky)
- EVANS, D. A.; NELSON J. V.; VOGEL E.; TABER T. R. Stereoselective aldol condensations via boron enolates. Journal of the American Chemical Society. 1981, s. 3099–3111. DOI 10.1021/ja00401a031. (anglicky)
- Evans D. A. Aldrichimica Acta 1982, 15, 23. (Review)
- Gage J. R.; Evans D. A., Diastereoselective Aldol Condensation Using A Chiral Oxazolidinone Auxiliary: (2S*,3S*)-3-Hydroxy-3-Phenyl-2-Methylpropanoic Acid Archivováno 29. 9. 2012 na Wayback Machine, Organic Syntheses, Coll. Vol. 8, p.339 (1993); Vol. 68, p.83 (1990).
- EVANS, D. A.; BARTROLI J.; SHIH T. L. Enantioselective aldol condensations. 2. Erythro-selective chiral aldol condensations via boron enolates. Journal of the American Chemical Society. 1981, s. 2127–2129. DOI 10.1021/ja00398a058. (anglicky)
- EVANS, D. A.; BENDER S. L.; MORRIS J. The total synthesis of the polyether antibiotic X-206. Journal of the American Chemical Society. 1988, s. 2506–2526. DOI 10.1021/ja00216a026. (anglicky)
- EVANS, D. A.; CLARK J.S.; METTERNICH R.; SHEPPARD G.S. Diastereoselective aldol reactions using .beta.-keto imide derived enolates. A versatile approach to the assemblage of polypropionate systems. Journal of the American Chemical Society. 1990, s. 866–868. DOI 10.1021/ja00158a056. (anglicky)
- EVANS, D. A.; NG, H.P.; CLARK, J.S.; RIEGER, D.L. Diastereoselective anti aldol reactions of chiral ethyl ketones. Enantioselective processes for the synthesis of polypropionate natural products. Tetrahedron. 1992, s. 2127–2142. DOI 10.1016/S0040-4020(01)88879-7. (anglicky)
- Shigehisa, H.; Mizutani, T.; Tosaki, S. Y.; Ohshima, T.; Shibasaki, M, Tetrahedron 2005, 61, 5057-5065.
- S. B. Jennifer Kan; KENNETH K.-H. NG; IAN PATERSON. The Impact of the Mukaiyama Aldol Reaction in Total Synthesis. Angewandte Chemie International Edition. 2013, s. 9097–9108. DOI 10.1002/anie.201303914. (anglicky)
- Teruaki Mukaiyama; KAZUO BANNO; KOICHI NARASAKA. Reactions of silyl enol ethers with carbonyl compounds activated by titanium tetrachloride. Journal of the American Chemical Society. 1974, s. 7503–7509. DOI 10.1021/ja00831a019. (anglicky)
- 3-Hydroxy-3-Methyl-1-Phenyl-1-Butanone by Crossed Aldol Reaction Teruaki Mukaiyama and Koichi Narasaka Organic Syntheses, Coll. Vol. 8, p.323 (1993); Vol. 65, p.6 (1987)
- Carreira E.M.; SINGER R.A.; LEE W.S. Catalytic, enantioselective aldol additions with methyl and ethyl acetate O-silyl enolates — a chira; tridentate chelate as a ligand for titanium(IV). Journal of the American Chemical Society. 1994, s. 8837–8. DOI 10.1021/ja00098a065. (anglicky)
- CARREIRA, E. M.; FETTES, A.; MARTL, C. Catalytic Enantioselective Aldol Addition Reactions. Organic Reactions. 2006, s. 1–216. ISBN 0471264180. DOI 10.1002/0471264180.or067.01. (anglicky)
- Z. G. Hajos, D. R. Parrish, German Patent DE 2102623 1971
- HAJOS, Zoltan G.; PARRISH, DAVID R. Asymmetric synthesis of bicyclic intermediates of natural product chemistry. Journal of Organic Chemistry. 1974, s. 1615–1621. DOI 10.1021/jo00925a003. (anglicky)
- EDER, Ulrich; SAUER, GERHARD; WIECHERT, RUDOLF. New Type of Asymmetric Cyclization to Optically Active Steroid CD Partial Structures. Angewandte Chemie International Edition in English. 1971, s. 1615–1621. DOI 10.1002/anie.197104961. (anglicky)
- LIST, Benjamin. The ying and yang of asymmetric aminocatalysis. Chemical Communications. 2006, s. 819–824. DOI 10.1039/b514296m. PMID 16479280. (anglicky)
Externí odkazy
 Obrázky, zvuky či videa k tématu Aldolová reakce na Wikimedia Commons
Obrázky, zvuky či videa k tématu Aldolová reakce na Wikimedia Commons