Stilleova reakce
Stilleova reakce je organická reakce, často používaná v organické syntéze. Spočívá v párování dvou organických sloučenin, přičemž jednou z nich je organocínová sloučenina (neboli organostannan). Druhou sloučeninou může být některý z mnoha organických elektrofilů. Stilleova reakce patří mezi křížové párovací reakce.[1][2][3]

- : Allyl, alkenyl, aryl, benzyl, acyl
- : halogenidy (Cl, Br, I), pseudohalogenidy (OTf, ), OAc



Skupina R1 navázaná na trialkylcín je obvykle sp2-hybridizovaná, může jít například o vinyl nebo aryl.
Organostannany jsou odolné vůči vzduchu i vodě, řada z nich je buď komerčně dostupná, nebo připravitelná podle postupu popsaného v literatuře; často ovšem tyto látky bývají značně toxické. X bývá halogenid, většinou Cl, Br nebo I, i když je možné použít i pseudohalogenidy, jako jsou trifláty, také sulfonáty a fosfáty.[4][5] Touto reakcí se zabývalo několik studií.[6][2][7][8][9][10][11][12][13][14][15]
Historie
První křížové párování arylhalogenidů za přítomnosti organocínových sloučenin popsal Colin Eaborn v roce 1976.[16] Tato reakce dávala diarylový produkt se 7% až 53% výtěžností. Tento proces roku 1977 rozšířil Tošihiko Migita o párování acylchloridů s alkylcínovými sloučeninami, přičemž se tvořil keton s výtěžností od 53 % do 87 % .[17]
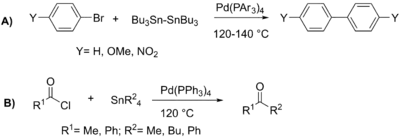
V roce 1977 T. Migita vydal práci zabývající se reakcemi allylcínových sloučenin s aryl- (C) a acylhalogenidy (D). Větší schopnost allylových skupin přesouvat se na palladiový katalyzátor umožnila provádět reakce za nižších teplot. Výtěžnosti se u arylhalogenidů pohybovaly od 4 do 100 %, a u acylhalogenidů mezi 27 a 86 %.[18][19] Na základě Migitových a Kosugiových objevů se pro Stilleovu reakci někdy používá označení Migitova–Kosugiova–Stilleova reakce.
John Kenneth Stille následně v roce 1978 provedl párování mnoha různých alkylcínových sloučenin s řadou aryl- a acylhalogenidfů za mírných podmínek a s mnohem lepšími výtěžnostmi (76-99 %).[18][20] V 80. letech Stille pokračoval v práci na syntéze mnoha ketonů pomocí tohoto univerzálního a mírného procesu a objasnil mechanismus této přeměny.[21][22]

K polovině 80- let 20. století bylo publikováno 65 prací zabývajících se párovacími reakcemi využívajícími organocínové sloučeniny, čímž se dále rozšiřovalo spektrum použitelných substrátů. Zatímco původní výzkum byl zaměřen na párování alkylových skupin, tak se další práce soustředily především na využitelnější párování vinylových, alkenylových, arylových a allylových organostannanů. Díky stabilitě organocínových sloučenin za přítomnosti vzduchu a jejich snadné přípravě se Stilleova reakce stala často využívaným postupem.[8]
Mechanismus
Mechanismus Stilleovy reakce byl podrobně prozkoumán.[11][23] Katalytický cyklus obsahuje oxidační adici halogenidu nebo pseudohalogenidu (2) na palladiový katalyzátor (1), transmetalaci meziproduktu 3 s organocínovým činidlem (4) a redukční eliminaci 5 za vzniku konečného produktu (7) a obnovení katalyzátoru (1).[24]
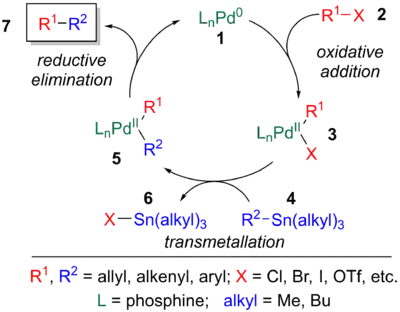
Přesný mechanismus Stilleova párování je složitý a může probíhat řadou způsobů. Podobně jako u jiných křížových párování katalyzovaných palladiem se předpokládá vznik 14elektronového komplexu Pd0, který se může vytvořit několika způsoby. 18- nebo 16elektronový zdroj Pd0, například Pd(PPh3)4 nebo Pd(dba)2, odštěpí ligand za tvorby aktivního katalyzátoru. V druhém kroku se fosfin naváže na volné Pd0. Může také proběhnout redukce zdroje PdII, (8) (což může být například (Pd(OAc)2, PdCl2(MeCN)2, PdCl2(PPh3)2 nebo BnPdCl(PPh3)2) navázáním fosfinového ligandu nebo organocínové sloučeniny.[6]
Oxidační adice
Oxidační adice se účastní 14elektronový komplex Pd0. Vzniká přitom 16elektronový meziprodukt obsahující PdII. Tento krok pravděpodobně urychlují aniontové ligandy, jako OAc, tvorbou [Pd(OAc)(PR3)n]−, což zvyšuje nukleofilitu komplexů palladia.[11][25]
V některých případech, například při použití sp3 hybridizovaných organohalogenidů, převažuje SN2 mechanismus.[11][25]
Přestože se po soustředěných oxidačních adicích obvykle vytváří cis-meziprodukt, tak rychle vzniká rovnováha mezi cis- a trans-izomerem.[26][27]
Isomerizace má několik příčin. První z nich spočívá v tom, že se obvykle na fosfiny navazují objemné skupiny, pro které je vzájemná orientace cis značně nevýhodná, čímž dochází k izomerizaci na trans-produkt.[26][27] Druhou příčinu lze vysvětlit pomocí sdn modelu.[24][28] Podle této teorie je palladium hypervalentní. R1 a trans ligand tak „soupeří“ o navázání na jeden orbital palladia. Vzniklá čtyřelektronová tricentrická vazba je nejslabší tehdy, když jsou přítomny dvě silně donorové skupiny, které o orbital soupeří silně. Ve srovnání s ostatními běžně používanými ligandy má C-donor R1 ligandu mnohem výraznější trans efekt. Ten ukazuje míru soupeření mezi ligandy, které jsou navzájem v poloze trans, o orbital palladia. Obvykle používané fosfiny a C-donory (R1) a patří mezi měkké ligandy, vytvářejí s palladiem silnější vazby, a výrazně soupeří o navázání.[29][30] Protože halogenidy a pseudohalogenidy jsou výrazně elektronegativnější, tak jsou jejich vazby s palladiem značně polarizované s větší elektronovou hustotou na skupině X, a tak vytvářejí mnohem slabší trans efekty. Z tohoto důvodu je pro R1 výrazně výhodnější být vůči X v poloze trans, protože R1 poté vytvoří silnější vazbu s palladiem.[24][28][30]
Transmetalace
Transmetalace trans produktu oxidační adice může probíhat několika různými mechanismy, které závisí na substrátech a na podmínkách reakce. Nejčastější je u Stilleova párování asociativní mechanismus, kdy se organocínová sloučenina, obvykle atom cínu navázaný na allylovou, alkenylovou nebo arylovou skupinu, koordinuje na palladium skrz jednu z dvojných vazeb. Tímto vzniká pětivazný 18elektronový meziprodukt, jenž poté odštěpí ligand za vzniku rovinného čtvercového komplexu. Přestože je organostannan na palladium koordinován přes skupinu R2, tak musí být R2 formálně přenesena na palladium (vazba R2-Sn musí zaniknout) a skupina X musí odejít společně s cínem, čímž se transmetalace dokončí. Tato část může probíhat dvěma způsoby.[31]
V prvním po navázání organostannanu na trans komplex dojde ke koordinaci skupiny X na cín a utvoří se cyklický přechodný stav. Rozpadem tohoto aduktu zanikne vazba R3Sn-X a trojvazný komplex má R1 a R2 v poloze cis.
Druhý častý mechanismus začíná stejnou adicí organostannanu na trans-komplex; skupina X se však nekoordinuje na cín, vzniklý přechodný stav tak má otevřený řetězec. Poté uhlík v poloze alfa vzhledem k cínu atakuje palladium, čímž komplex cínu získá celkový kladný náboj. Na níže uvedeném schématu dvojná vazba koordinující se na cín odpovídá R2, tedy alkenylu, allylu nebo arylu. X může kdykoliv disociovat a navázat se na Sn+ komplex. Výpočty založené na teorii funkcionálu hustoty předpovídají, že otevřený mechanismus bude převládat, jestliže dva ligandy zůstanou navázané na palladium a skupina X se oddělí, cyklický proběhne přednostně, když bude ligand disociovat před transmetalací. Dobré odstupující skupiny, jako jsou (v polárních rozpouštědlech) trifláty tak upřednostňují první mechanismus, zatímco u objemných fosfinových ligandů lze očekávat převahu druhého.[31]
Méně častý bývá disociativní mechanismus, kde ligand ze čtyřvazné sloučeniny palladia disociuje a koordinující rozpouštědlo se může navázat na palladium. Po odštěpení rozpouštědla se vytvoří 14elektronový trojvazný meziprodukt a organostannan se může navázat na palladium, po čemž může proběhnout cyklický nebo otevřený proces, jak je popsáno výše.[31]
Redukční eliminace
Aby mohlo dojít k redukční eliminaci R1-R2, tak tyto skupiny musí být vzájemně v poloze cis. Jakýkoliv trans-adukt se tak musí izomerizovat na cis meziprodukt, jinak nebude dále reagovat. Redukční eliminace může proběhnout několika způsoby, většinou jde o soustředěné mechanismy.[11][32][33]
Nejprve 16elektronový čtyřvazný meziprodukt vzniklý transmetalací podstoupí redukční eliminaci ze čtvercového rovinného komplexu. Tato část probíhá ve dvou krocích: nejprve po redukční eliminaci proběhne eliminace a následně koordinace nově utvořené vazby sigma mezi R1 a R2 na kov, čímž se utvoří výsledný produkt.[11][32][33]
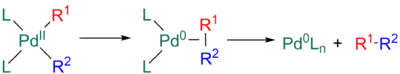
Tento proces ovšem bývá někdy pomalý, může být výrazně urychlen disociací ligandu za tvorby 14elektronového meziproduktu ve tvaru T. Tento meziprodukt se poté může přesmyknout do tvaru písmene Y, kde je redukční eliminace rychlejší.[11][32][33]
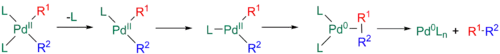
Nakonec se může další ligand navázat na palladium za vzniku 18elektronové trigonálně bipyramidové struktury, přičemž R1 a R2 jsou ekvatoriální a navzájem v poloze cis. Geometrie tohoto meziproduktu je podobná jako u předchozího ve tvaru Y.[11][32][33]
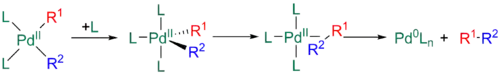
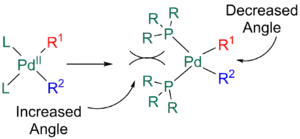
Rychlost eliminace mohou též navýšit objemné ligandy, například fosfiny s velkými hodnotami úhlu ligand-kov-ligand způsobují sterické odpuzování L s R1 a R2, čímž se zvětšuje úhel mezi L a R a úhel mezi R1 a R2 tak klesá, díky čemuž je redukční eliminace rychlejší.[11][24]
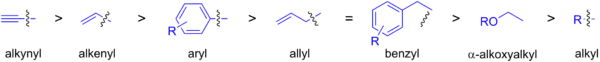
Kinetika
Níže jsou zobrazeny rychlosti transmetalací různých organostannanů. Nejčastějšími párovacími partnery jsou zde sp2-hybridizované skupiny navázané na cín, jelikož sp3-hybridizované uhlíky vyžadují tvrdší reakční podmínky a koncové alkyny lze párovat přes vazby C-H v Sonogaširových reakcích.
Nejčastějšími organocínovými reaktanty bývají trimethylstannylové a tributylstannylové sloučeniny. Ačkoli jsou trimethylstannyly reaktivnější než odpovídající tributylstannyly a mají jednodušší 1H-NMR spektra, tak vykazují daleko vyšší toxicitu.[34]
Optimalizace výběru ligandů podle výtěžnosti a obratového čísla může být obtížná., protože oxidační adice vyžaduje kov bohatý na elektrony, ovšem upřednostňuje ligandy dodávající elektrony. Kovy chudší na elektrony jsou ovšem k transmetalacím a redukčním eliminacím náchylnější, takže jsou vhodnější ligandy, které elektronovou hustotu snižují. Nejvhodnější soustava ligandů tak záleží na použitých substrátech a reakčních podmínkách. Mohou se objevit změny v kroku určujícím rychlost, stejně jako v mechanismu transmetalace.[35]
Obvykle se používají ligandy se střední donační schopností, například fosfiny. K urychlení reakce lze použít ligandy s mírným nedostatkem elektronů, jako jsou tri-2-furylfosfin a trifenylarsenin. Ligandy se silnou donační schopností mohou párovací reakce zpomalovat či zcela inhibovat.[35][36]
Tato pozorování naznačují, že krokem určujícím rychlost Stilleovy reakce je většinou transmetalace.[36]
Přídavné látky
Látkou nejčastěji přidávanou ke Stilleovým reakcím, a to v katalytických i stechiometrických množstvích, je jodid měďný, jenž může reakci urychlit více než 103krát. Předpokládá se, že v polárních rozpouštědlech se měď transmetaluje s organostannanem. Vzniklé Gilmanovo činidlo se následně transmetaluje s palladiovým katalyzátorem. V etherových rozpouštědlech měď též urychluje odštěpení fosfinového ligandu, čímž aktivuje Pd centrum.[9][37][38][39][40]
Chlorid lithný urychluje Stilleovy reakce, při kterých použité skupiny X disociují od palladia (takže reakce probíhá mechanismem). Chloridový ion může oddělit X od palladia a tím zvýšit jeho aktivitu vůči transmetalaci, nebo se koordinovat na Pd0 adukt, přičemž v obou případech navyšuje rychlost oxidační adice. LiCl také vylepšuje polaritu rozpouštědla, což jinak aniontovým ligandům (–Cl, –Br, –OTf...) usnadňuje odštěpení. Je nezbytný, pokud se jako rozpouštědlo používá tetrahydrofuran; pomocí polárnějších rozpouštědel, jako je N-methyl-2-pyrrolidon (NMP) jeho přidání není potřeba.
V případech, kdy transmetalace probíhá cyklickým mechanismem, může chlorid lithný průběh reakce zpomalit, protože v cyklickém mechanismu neutrální ligandy, například fosfiny, disociují namísto aniontové skupiny X.[10][41]
Zdroje fluoridových iontů, například fluorid cesný, také mohou ovlivnit katalytický cyklus. Fluorid urychluje reakce organotriflátů, nejspíše stejným způsobem jako chlorid lithný.[39]
Vedlejší reakce
Nejčastější vedlejší reakcí u Stilleových párování je homopárování stannanů za vzniku dimerů R2-R2, u níž byly navrženy dva mechanismy. U prvního reakce dvou ekvivalentů organostannanu s palladnatým prekatalyzátorem vytvoří homopárovaný produkt po redukční eliminaci. V druhém případě Pd0 vstupuje do radikálových pochodů. Organostannany, které se zde používají obvykle mají čtyřvazný cín, kde se vyskytuje sp2-hybridizovaná skupina a tři „nepřesunutelné“ alkyly. Alkyly se obvykle na palladium přesouvají nejpomaleji.[10]

Bylo zjištěno, že i při teplotách okolo 50 °C se mohou vyměnit arylové skupiny na palladiu a koordinovaném fosfinu. I když obvykle nejsou zaznamenány, tak mnohdy mají potenciál vytvořit menšinové produkty.[10]
Další, vzácnější, vedlejší reakcí je cine substituce. Po úvodní oxidační adici arylhalogenidu se mohou Pd-Ar meziprodukty navázat na dvojné vinyl-cínové vazby. Po následné beta-hydridové eliminaci, migrační inserci a protodestannylaci se tak může vytvořit 1,2-disubstituovaný alken.[10]
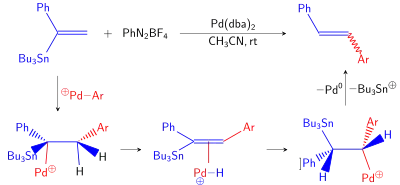
Existuje i řada dalších vedlejších reakcí narušujících Stilleova párování, jako jsou cis-trans izomerizace, jež narušují reakci po tvorbě alkenylstannanu. Mechanismus této přeměny není znám. Organostannany obvykle odolávají hydrolýzám, avšak u arylstannanů velmi bohatých na elektrony může jít o významnou vedlejší reakci.[10]
Přehled
Elektrofily
Častými párovacími partnery jsou u Stilleových reakcí vinylhalogenidy; jejich reakce jsou součástmi totálních syntéz řady přírodních látek. Nejobvykleji se používají vinyljodidy a vinylbromidy. Vinylchloridy nemívají dostatečnou reaktivitu při oxidačních adicích na Pd0. Většinou se do reakce zapojují jodidy: reagují rychleji a za mírnějších podmínek než bromidy. Tento rozdíl lze ukázat na párování vinyljodidu za přítomnosti vinylbromidu.[10]
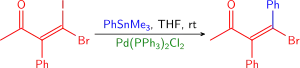
Stereochemie alkenu je v průběhu reakce obvykle zachována, s výjimkou tvrdších reakčních podmínek. Lze použít řadu různých alkenů, a to jak α- a β-halogen-α,β nenasycené ketony, estery a sulfoxidy (které obvykle vyžadují přídavek měďné soli) i mnoho dalších (viz následující obrázek).[42] Použít se někdy dají i vinyltrifláty. V některých případech je nutné přidání chloridu lithného (LiCl).[10]

Dalšími běžnými elektrofily jsou aryl- a heteroarylhalogenidy. Podobně jako u vinylových substrátů jsou bromidy a jodidy používány častěji. Spektrum použitelných arylových skupin je široké, patří sem kruhy se substituenty dodávajícími elektrony, biaryly a řada dalších. Jednou z možností jsou i halogenované heterocyklické sloučeniny, například pyridiny, furany, thiofeny, thiazoly, indoly, imidazoly, puriny, uracily, cytosiny a pyrimidiny.[10]
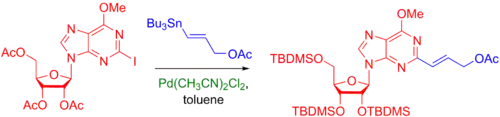
Níže jsou uvedeny příklady Stilleova párování při vytváření složitých struktur na nukleosidech, například purinech.[43]
Aryltrifláty a arylsulfonáty mohou být také párovány s velkým počtem organostannanů. Trifláty ve Stilleových reakcích reagují podobně jako bromidy.[10]
Acylchloridy mohou reagovat s organostannany, i alkylcínovými sloučeninami, za vzniku ketonů (viz níže).[44] Často je však obtížné navázat acylchloridy na velké molekuly obsahující citlivé funkční skupiny. Byla vyvinuta alternativa tohoto procesu, nazývaná Stilleova karbonylační párovací reakce, při níž dochází k napojení karbonylových skupin migrační insercí oxidu uhelnatého.[10]
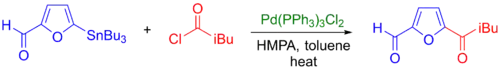
Do Stilleových reakcí je možné zapojit i allyl-, benzyl- a propargylhalogenidy. Allylhalogenidy reagují přes η3 přechodné stavy, což umožňuje párování s organostannany na pozicích α i γ, přičemž převážně probíhají na nejméně substituovaném atomu uhlíku.[45] Alkenylepoxidy (epoxidy s napojenými alkenylovými skupinami) mohou podstoupit stejný druh párování přes η3 přechodné stavy, přičemž se epoxidový kruh otvírá za tvorby alkoholu. Zatímco se allyl- a benzylacetáty používají často, tak propargylacetáty s organostannany nereagují.[10]

Stannany
Některé organostannany jsou komerčně dostupné.[46] Připravit je lze reakcemi Grignardových nebo organolithných činidel s trialkylcínchloridy, například vinyltributylcín se získává reakcí vinylmagnesiumbromidu s tributylcínchloridem.[47] Hydrostannylacemi alkynů či alkenů lze vytvořit velké množství těchto sloučenin. Organocínová činidla jsou odolná vůči vzduchu a vlhkosti. některé reakce dokonce mohou probíhat ve vodě.[48] Přečištění lze provést pomocí chromatografie. Některé organocínové sloučeniny, obzvláště trimethylstannyly, jsou toxické.[10]
Vinylstannany a další alkenylstannany jsou široce používány.[10] Stannany se značně objemnými substituenty nebo substitucemi na α-uhlících reagují pomalu, například α-substituovaný vinylstannan vstupuje pouze do reakcí s koncovými jodidy.[49]

Arylstannany jsou také běžné, transmetalace u nich urychlují jak substituenty. které dodávají elektrony, tak i ty, jež je odtahují, což naznačuje dva různé mechanismy transmetalace. Jediným omezením těchto reaktantů jsou substituenty v polohách ortho, kde i malé skupiny, jako jsou methyly, snižují rychlost reakce. K párování je možné použít i mnoho heterocyklů (jako je níže znázorněný příklad u thiazolu).[10][50]

I když jsou alkyly na organostannanech obvykle považovány za nepřesunutelné ligandy, tak jsou popsány případy, kdy takovéto skupiny, obzvláště benzyly, mohou být za vyšších teplot párovány. Pokud je na cín navázáno několik různých alkylových skupin, selektivita může být snížená. Alkylový párovací partner se musí přesouvat na palladium rychleji než „nepřesunutelný“ logand.[10][51] Tento problém lze vyřešit přípravou a použitím alkylkarbastannatranů.

Alkynylstannany, jež jsou z organostannanů nejreaktivnější, mohou rovněž být zapojeny do Stilleových reakcí. Nepoužívají se však často, protože koncové alkynovy mohou reagovat přímo s palladiem v Sonogaširových reakcích. Allylstannany reagují s obtížemi, podobně jako allylhalogenidy, protože se u nich obtížně řídí regioselektivitra α a γ adicí.
Jako reaktanty ve Stilleových reakcích lze použít rovněž distannany a acylstannany.[10]
Použití
Stilleovy reakce jsou zapojovány do výroby některých polymerů.[52][53][54] Největší využití ovšem mají v organické syntéze, hlavně při přípravách přírodních látek.
Totální syntézy přírodních látek
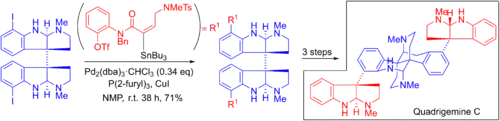
Larry Overman vyvinul 19krokovou enantioselektivní totální syntézu kvadrigeminu C zahrnující dvojitou Stilleovu metatezní reakci.[6][55] Komplexní organostannan je zde párován s dvěma aryljodidy. Po dvojité Heckově cyklizaci vzniká konečný produkt.
Panekova 32kroková enantioselektivní totální syntéza ansamycinového antibiotika (+)-mykotrienolu využívá tandemové Stilleovo párování makrocyklů. Organostannan zde má dvě koncové tributylcínové skupiny napojené na alken. Tento organostannan k sobě „přitiskne“ dva konce lineárního reaktantu za vzniku makrocyklu, čímž do procesu zapojí dvě chybějící methylenové jednotky. Po oxidaci aromatického jádra dusičnanem amonno-ceričitým následuje oddělení chránicí skupiny za vzniku výsledného produktu s 54% výtěžností těchto 3 kroků.[6][56]

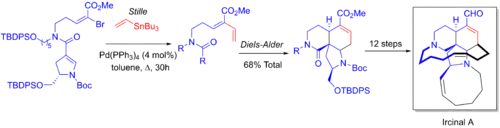
Stephen F. Martin vyvinul se svými spolupracovníky 21krokovou enantioselektivní syntézu manzaminového protinádorového alkaloidu ircinalu A s využitím jednonádobové kombinace Stilleovy a Dielsovy–Alderovy reakce. Alken se zde aduje na vinylbromid, poté probíhá in situ Dielsova–Alderova cykloadiční reakce adovaného alkenů s alkenovou skupinou pyrrolidinu.[6][57]
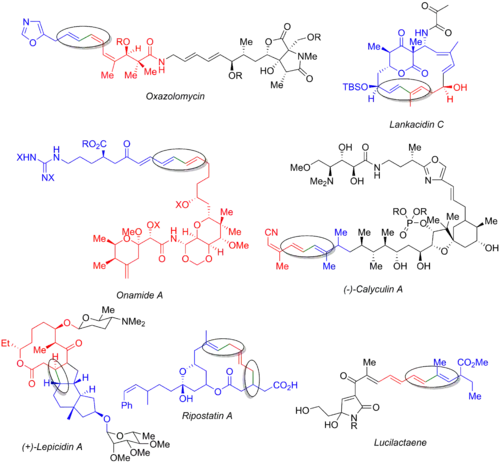
Je popsána řada dalších totálních syntéz využívajících Stilleovy reakce, například u oxazolomycinu,[58] lankacidinu C,[59] onamidu A,[60] kalyukulinu A,[61] lepicidinu A,[62] ripostatinu A,[63] a lucilaktaenu.[6][64] Níže uvedený obrázek znázorňuje výsledný produkt, organohaogenid (modře), organostannan (červeně) a vytvořenou vazbu (zeleně, v kruhu).Z těchto příkladů je zřejmé, že Stilleovy reakce lze použít v úvodních částech syntéz (oxazolomycin and calykulin A), ke konci procesu (onamid A, lankacidin C, ripostatin A), nebo přibližně uprostřed (lepicidin A a lucilaktaen). Při syntéze ripostatinu A probíhají současně dvě Stilleova párování, po kterých následuje uzavírací metateze. Syntéza lucilaktaenu obsahuje skupinu s boranem na jedné straně a stannanem na druhé, což po Stilleově párování umožňuje provést Suzukiovu reakci.
Obměny
Za účelem vylepšení reakce byla vyzkoušena řada různých organických rozpouštědel, byly také vyvinuty Stilleovy reakce ve vodných roztocích.[14]
Za přítomnosti měďných solí se jako účinný katalyzátor ukázalo palladium- na uhlíku.[65][66]
V zelené chemii Stilleovy reakce probíhají v nízkotajících vysoce polárních směsích cukrů, jako například mannitolu, derivátů močoviny, například dimethylmočoviny a solí (jako je chlorid amonný)[67][68] Katalytický systém je tvořen tris(dibenzylidenaceton)dipalladiem a trifenylarsinem:

Stilleovo karbonylační párování
Běžnou obdobou Stilleova párování je zavedení karbonylové skupiny mezi R1 a R2, což představuje účinný způsob přípravy ketonů. Tento proces je velmi podobný párování organostannanů s acylchloridy. Tyto skupiny však nejsou vždy dostupné a jejich příprava může být těžká, obzvláště, pokud jsou přítomny citlivé funkční skupiny. Obtížné je někdy i ovládání jejich reaktivity. Stilleova karbonylační párování se provádějí za stejných podmínek jako klasické Stilleovy reakce, pouze atmosféra je tvořena oxidem uhelnatým (CO). CO se může koordinovat na R1 palladiového katalyzátoru (9) po úvodní oxidační adici, jež je následována navázáním CO na vazbu Pd-R1 bond (10), což pak vede k redukční eliminaci za tvorby ketonu (12). Krokem určujícím rychlost reakce je zde většinou transmetalace.[6]

Larry Overman se svými spolupracovníky zapracoval Stilleovo karbonylační párování do dvacetikrokové enantioselektivní totální syntézy strychninu. Přidaný karbonyl byl později přeměněn na alken ve Wittigově reakci, což umožnilo tvorbu potřebného terciárního dusíku a pětičlenného cyklického jádra kombinací aza-Copeova přesmyku s Mannichovou reakcí.[6][69]

Giorgio Ortar et al. využili Stilleovu karbonylační párovací reakci na syntézu benzofenonfosforů. Ty poté byly vloženy do 4-benzoyl-L-fenylalaninových peptidů, jenž byly následně použity ke zkoumání interakcí peptidů s bílkovinami.[6][70]
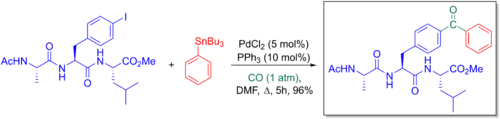
Louis Hegedus vyvinul šestnáctikrokovou přípravu racemického jatrafonu, jejíž poslední součástí byla Stilleova karbonylační reakce vytvářející jedenáctičlenný cyklus. Namísto halogenidu zde byl použit vinyltriflát.[6][71]
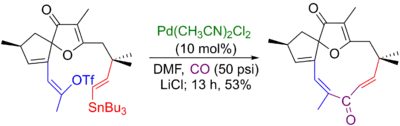
Stilleovo–Kellyovo párování
Na základě práce Colina Eaborna z roku 1976, popisující tvorbu arylstannanů z arylhalogenidů a distannanů, T. Ross Kelly našel způsob vnitromolekulárního párování arylhalogenidů. Toto spojení stannylace s párováním arylhalogenidů bylo použito na přípravu několika dihydrofenantrenů. Tímto postupem se většinou dají získat 5členné nebo 6členné kruhy, byly však připraveny i makrocykly. Na rozdíl od běžné Stilleovy reakce zde chlor nefunguje stejně jako ostatní halogeny., pravděpodobně v důsledku své nižší reaktivity (vytváří kratší a silnější vazby, které se při oxidačních adicích hůře štěpí).
Na začátku se palladiový katalyzátor (1) oxidačně naaduje na nejreaktivnější vazbu C-X (13) za vzniku meziproduktu 14, poté proběhne transmetalace s distannanem (15) za tvorby 16 a redukční eliminace, která vede ke tvorbě arylstannanu (18). Obnovený katalyzátor (1) se může navázat na druhou vazbu C-X bond sloučeniny 18, čímž se utvoří 19, z něhož vnitromolekulární transmetalací vznikne meziprodukt 20, jenž je redukčně eliminován na (22).[6]

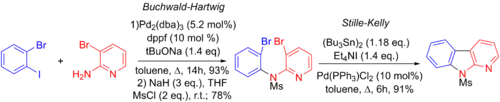
Stilleova-Kellyova reakce byla též použita na přípravu několika benzo[4,5]furopyridinů. Jedná se o třífázový proces, skládající se z Buchwaldovy–Hartwigovy aminace, dalšího křížového párování a vnitromolekulární Stilleovy-Kellyovy reakce. Vazba mezi arylem a jodidem se na palladium aduje rychleji než vazba aryl-bromid.[6][72]
Odkazy
Související články
- Organocínové sloučeniny
- Křížová párovací reakce
- Suzukiova reakce
- Negišiovo párování
- Heckova reakce
- Hijamovo párování
Externí odkazy
Reference
V tomto článku byl použit překlad textu z článku Stille reaction na anglické Wikipedii.
- Hartwig, J. F. Organotransition Metal Chemistry, from Bonding to Catalysis; University Science Books: New York, 2010. ISBN 189138953X
- Stille, J. K. Angewandte Chemie International Edition in English 1986, 25, 508–524. (Review)
- Farina, V.; Krishnamurthy, V.; Scott, W. J. Organic Reactions 1998, 50, 1–652. (Review)
- Scott, W. J.; Crisp, G. T.; Stille, J. K. Organic Syntheses, Coll. Vol. 8, p. 97 (1993); Vol. 68, p. 116 (1990). (Article)
- Stille, J. K.; Echavarren, A. M.; Williams, R. M.; Hendrix, J. A. Organic Syntheses, Coll. Vol. 9, p.553 (1998); Vol. 71, p.97 (1993). (Article)
- Kurti, L.; Czako, B. Strategic Applications of Named Reactions in Organic Synthesis; Elsevier: Burlington, 2005.
- Mitchell, T. N. J. Organomet. Chem., 1986, 304, 1-16.
- Mitchell, T. N. Synthesis, 1992, 803-815. (DOI:10.1055/s-1992-26230)
- Farina, V. Pure Appl. Chem., 1996, 68, 73–78. (DOI:10.1351/pac199668010073).
- Farina, V.; Krishnamurthy, V.; Scott, W. J. The Stille Reaction; Wiley: Online, 2004. (DOI:10.1002/0471264180.or050.01).
- Espinet, P.; Echavarren, A. M. Angew. Chem. Int. Ed., 2004, 43, 4704–4734.(DOI:10.1002/anie.200300638)
- Pattenden, G.; Sinclair, D. J. J.Organomet. Chem., 2002, 653, 261-268.
- Kosugi, M.; Fugami, K. J. Organomet. Chem., 2002, 19, 10-16.
- Pierre Genet, J.; Savignac, M. J. Organomet. Chem., 1999, 576, 305-317.
- Cordova, C.; Bartolomé, C.; Martínez-Ilarduya, J.M..; Espinet, P. ACS Catal., 2015, 5, 3040–3053.(DOI:10.1021/acscatal.5b00448)
- Azarian, D.; Dua, S. S.; Eaborn, C.; Walton, D. R. M. J. Organomet. Chem., 1976, 117, C55-C57. (DOI:10.1016/S0022-328X(00)91902-8)
- Kosugi, M.; Shimizu, Y.; Migita, T. Chemistry Letters, 1977, 6, 1423-1424. (DOI:10.1246/cl.1977.1423)
- Kosugi, M.; Sasazawa, K.; Shikizu, Y.; Migita, T. Chemistry Letters, 1977, 6, 301-302. (DOI:10.1246/cl.1977.301)
- Kosugi, M.; Shimizu, Y.; Migita, T. J. Organomet. Chem., 1977, 129, C36-C38. (DOI:10.1016/S0022-328X(00)92505-1)
- Milstein, D.; Stille, J. K. Journal of the American Chemical Society, 1978, 100, 3636-3638. (DOI:10.1021/ja00479a077)
- Milstein, D.; Stille, J. K. Journal of the American Chemical Society, 1979, 101, 4992-4998. (DOI:10.1021/ja00511a032)
- Milstein, D.; Stille, J. K. The Journal of Organic Chemistry, 1979, 44, 1613-1618. (DOI:10.1021/jo01324a006)
- Casado, A. L.; Espinet, P.; Gallego, A. M. Journal of the American Chemical Society, 2000, 122, 11771-11782. (DOI:10.1021/ja001511o)
- Crabtree, R. H. The Organometallic Chemistry of the Transition Metals, 5th ed.; Wiley: New York, 2009.
- Perez-Temprano, M. H.; Gallego, A. M.; Casares, J. A.; Espinet, P. Organometallics, 2011, 30, 611-617. (DOI:10.1021/om100978w)
- Minniti, D. Inorg. Chem, 1994, 33, 2631-2634.(DOI:10.1021/ic00090a025)
- Casado, A. L.; Espinet, P. Organometallics, 1998, 17, 954-959. (DOI:10.1021/om9709502)
- Landis, C. R.; Firman, T. K.' Root, D. M.; Cleveland, T. Journal of the American Chemical Society, 1998, 120, 1842-1854. (DOI:10.1021/ja9710114).
- Vicente, J.; Arcas, A.; Bautista, D. Organometallics, 1997, 16, 2127-2138. (DOI:10.1021/om961094h).
- Pearson, R. G. Inorg. Chem, 1973, 12, 712-713.(DOI:10.1021/ic50121a052)
- Garcia-Melchor, M.; Braga, A. A. C.; Lledos, A.; Ujaque, G.; Maseras, F. Acc. Chem. Res., 2013, 46, 2626-2634. (DOI:10.1021/ar400080r)
- Gillie, A.; Stille, J. K. Journal of the American Chemical Society, 1980, 102, 4933-4941. (DOI:10.1021/ja00535a018).
- Brown, J. M.; Cooley, N. A. Chemical Reviews, 1988, 88, 1031-1046. (DOI:10.1021/cr00089a003).
- McKillop, A.; Abel, E. W.; Stone, F. G. A.; Wilkinson, G. Comprehensive Organometallic Chemistry II, Elsevier Scientific: Oxford, 1995.
- Farina, V.; Journal of the American Chemical Society, 1991, 113, 9585-9595. (DOI:10.1021/ja00025a025).
- http://hwpi.harvard.edu/files/myers/files/11-the_stille_reaction.pdf
- Liebeskind, L. S.; Fengl, R. W. The Journal of Organic Chemistry, 1990, 55, 5359-5364. (DOI:10.1021/jo00306a012).
- Farina, V.; Kapadia, S.; Brishnan, B.; Wang, C.; Liebeskind, L. S. JThe Journal of Organic Chemistry, 1994, 59, 5905-5911. (DOI:10.1021/jo00099a018).
- Mee, S. P. H.; Lee, V.; Baldwin, J. E. Angewandte Chemie International Edition., 2004, 43, 1132-1136.
- Liebeskind, L. S.; Peña-Cabrera, E. Organic Syntheses, Coll. Vol. 10, p.9 (2004); Vol. 77, p.135 (2000). (Article)
- Scott, W. J.; Stille, J. K. Journal of the American Chemical Society, 1986, 108, 3033-3040. (DOI:10.1021/ja00271a037).
- Johnson, C. R.; Adams, J. P.; Braun, M.P.; Senanayake, C. B. W. Tetrahedron Lett., 1992, 33, 919-922. (DOI:10.1016/S0040-4039(00)91576-4)
- Nair, V.; Turner, G. A.; Chamberlain, S. D. Journal of the American Chemical Society, 1987, 109, 7223-7224. (DOI:10.1021/ja00257a071)
- Jousseaume, B.; Kwon, W.; Verlhac, J. B.; Denat, F.; Dubac, J. Synlett, 1993, 117-118. (DOI:10.1055/s-1993-22368)
- Sheffy, F. K.; Godschalx, J. P.; Stille, J. K. Journal of the American Chemical Society, 1984, 106, 4833-4840. (DOI:10.1021/ja00329a032)
- http://www.sigmaaldrich.com/chemistry/chemistry-products.html?TablePage=16246425
- Dietmar Seyferth. Di-n-butyldivinyltin. Organic Syntheses. 1959, s. 10. DOI 10.15227/orgsyn.039.0010. (anglicky)
- Wolf, C.; Lerebours, R. J. Org. Chem., 2003,68 7551-7554. (DOI:10.1021/jo0347056).
- Crisp, G.T.; Glink, P. T. Tetrahedron, 1994, 50, 2623. (DOI:10.1016/S0040-4020(01)86978-7)
- Bailey, T. R. Tetrahedron Lett., 1986, 27, 4407. (DOI:10.1016/S0040-4039(00)84964-3)
- Nativi, C.; Ricci, A.; Taddei, M. Tetrahedron Letters, 1990, 31, 2637. (DOI:10.1016/0040-4039(90)80147-E).
- Bao, Z.; Chan, W.; Yu, L. Chemistry of Materials, 1993, 5, 2-3. (DOI:10.1021/cm00025a001).
- Bao, Z.; Chan, W. K.; Yu, L. Journal of the American Chemical Society, 1995, 117, 12426-12435. (DOI:10.1021/ja00155a007)
- Sun, S. S.; Lewis, J. E.; Zhang, J.; Jiang, X.; Zhang, C.; Matos, T.; Li, R.; 'Polymer Chemistry, 2010, 1, 663-669. (DOI:10.1039/B9PY00324J)
- Lebsack, A. D.; Link, J. T.; Overman, L. E.; Stearns, B. A. Journal of the American Chemical Society, 2002, 124, 9008-9009. (DOI:10.1021/ja0267425)
- Masse, C. E.; Yang, M.; Solomon, J.; Panek, J. S. Journal of the American Chemical Society, 1998, 120, 4123-4134. (DOI:10.1021/ja9743194)
- Martin, S. F.; Humphrey, J. M.; Ali, A.; Hillier, M. C. Journal of the American Chemical Society, 1999, 121, 866-867. (DOI:10.1021/ja9829259)
- Kende, A. S.; Kawamura, K.; DeVita, R. J. Journal of the American Chemical Society, 1990, 112 4070-4072. (DOI:10.1021/ja00166a072)
- Kende, A. S., Koch, K.; Dorey, G.; Kaldor, I.; Liu, K. Journal of the American Chemical Society, 1993, 115, 9842-9843. (DOI:10.1021/ja00074a078)
- Hong, C. Y, Kishi, Y. Journal of the American Chemical Society, 1991, 113, 9693-9694. (DOI:10.1021/ja00025a056)
- Tanimoto, N.; Gerritz, S. W.; Sawabe, A.; Noda, T.; Filla, S. A.; Masamune, S. Angewandte Chemie International Edition, 2003, 33, 673-675. (DOI:10.1002/anie.199406731)
- Evans, D. A.; Black, W. C. Journal of the American Chemical Society, 1993, 115, 4497-4513. (DOI:10.1021/ja00064a011)
- Tang, W.; Prusov, E. V. Organic Letters, 2012, 14 4690-4693. (DOI:10.1021/ol302219x)
- Coleman, R. S.; Walczak, M. C.; Campbell, E. L. Journal of the American Chemical Society, 2005, 127, 16036-16039. (DOI:10.1021/ja056217g)
- Roth, G. P.; Farina, V.; Liebeskind, L. S.; Peña-Cabrera, E. Tetrahedron Letters 1995, 36, 2191.
- Renaldo, A. F.; Labadie, J. W.; Stille, J. K. Organic Syntheses, Coll. Vol. 8, p. 268 (1993); Vol. 67, p.86 (1989). (Article)
- Stille Reactions with Tetraalkylstannanes and Phenyltrialkylstannanes in Low Melting Sugar-Urea-Salt MixturesGiovanni Imperato, Rudolf Vasold, Burkhard König Advanced Synthesis & Catalysis Volume 348, Issue 15 , Pages 2243–47 2006 DOI:10.1002/adsc.2006
- P. Espinet, A. M. Echavarren. The Mechanisms of the Stille Reaction. Angewandte Chemie International Edition. 2004, s. 4704–4734. DOI 10.1002/anie.200300638. PMID 15366073. (anglicky)
- Knight, S. D.; Overman, L. E.; Pairaudeau, G. Journal of the American Chemical Society, 1993, 115, 9293-9294. (DOI:10.1021/ja00073a057)
- Monera, E.; Ortar, G. Bioorganic & Medicinal Chemistry Letters, 2000, 10, 1815-1818. (DOI:10.1016/S0960-894X(00)00344-9)
- Gyorkos, A. C.; Stille, J. K.; Hegedus, L. S. Journal of the American Chemical Society, 1990, 112, 8465-8472. (DOI:10.1021/ja00179a035)
- YUE, Wen Song; LI, Jie Jack. A Concise Synthesis of All Four Possible Benzo[4,5]furopyridines via Palladium-Mediated Reactions. Organic Letters. 2002-06-01, roč. 4, čís. 13, s. 2201–2203. Dostupné online [cit. 2021-11-05]. ISSN 1523-7060. DOI 10.1021/ol0260425. (anglicky)