Dielsova–Alderova reakce
Dielsova–Alderova reakce je reakce konjugovaného dienu s alkenem za vzniku cyklohexenu nebo jeho derivátu; jedná se o zvláštní příklad pericyklické reakce, která se podrobněji klasifikuje jako tepelně přípustná [4+2] cykloadice s Woodwardovým–Hoffmannovým symbolem [π4s + π2s]. Popsali ji Otto Diels a Kurt Alder roku 1928 a roku 1950 obdrželi za tento objev Nobelovu cenu za chemii. Díky tvorbě nových vazeb uhlík-uhlík představuje užitečný postup přípravy šestičlenných cyklů, přičemž se dá dobře ovládat regioselektivita a stereoselektivita vznikajících produktů.[1][2] Lze ji využít k získání složitějších struktur při výrobě mnoha látek.[3] Dielsovu–Alderovu reakci lze použít i u π-systémů s heteroatomy, jako jsou karbonylové sloučeniny a iminy, pak se vytváří příslušné heterocyklické sloučeniny. Reakci lze provést i k přípravě cyklů s jiným počtem atomů, zde ovšem nedosahuje takové univerzálnosti.
Vzhledem k záporným hodnotám ΔH° a ΔS° u běžných Dielsových–Alderových reakcí je provedení zpětné reakce snazší při vyšších teplotách; takové reakce se však využívají jen zřídka, zejména u Dielsových–Alderových adduktů s určitou zvláštní strukturou.[4]
Mechanismus
Dielsova–Alderova reakce patří mezi soustředěné pericyklické reakce.[5] Předpokládá se, že během ní vzniká jediný, cyklický, meziprodukt.[6] Klasifikuje se jako [π4s + π2s] cykloadice, což znamená, že jde o suprafaciální/suprafaciální interakci systému čtyř π elektronů (což odpovídá struktuře dienu) se systémem 2 π elektronů (strukturou dienofilu), interakce, při které vzniká přechodný stav bez energetické bariéry zapříčiněné orbitalovou symetrií a která tak umožňuje poměrně snadný průběh reakce.[7]
Při reakci dochází k vzájemnému působení HOMO/LUMO mezi ψ2 na elektrony bohatého dienu, stejně jako u na elektrony chudého dienofilu. Rozdíl energetické hladiny HOMO a LUMO je zde však tak malý, že může dojít k prohození těchto procesů. Při obrácené Dielsově–Alderově reakci substituenty na dienu, které snižují elektronovou hustotu, snižují energii jeho prázdných orbitalu ψ3 a substituenty dodávající elektrony zvyšují energii orbitalu π do takové míry, že se interakce mezi těmito orbitaly stávají nejvýznamnějšími stabilizujícími orbitalovými interakcemi; HOMO a LUMO reaktantů se tak nacházejí ve stavu, kdy dochází k tvorbě vazeb, jak je zobrazeno níže. Jelikož se reaktanty nacházejí ve svých základních stavech, tak k zahájení reakce stačí teplo a není třeba dodávat energii pomocí světla.[7]
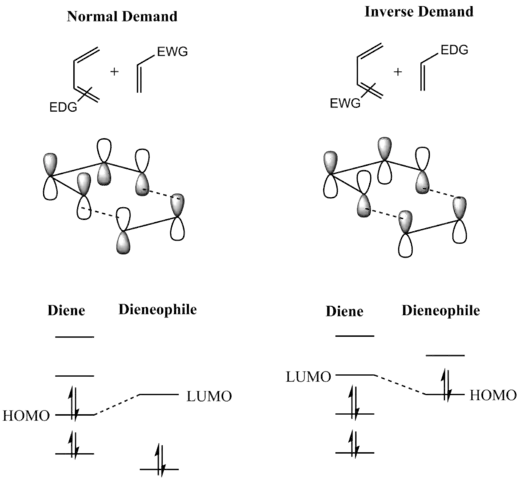
„Převažující domněnkou“ je, že takto probíhá většina Dielsových–Alderových reakcí;[8][9] [10][11] což je však dosud předmětem sporů. I když se většina Dielsových–Alderových reakcí ukazuje jako stereospecifické syn adice dvou látek, tak byl navržen i diradikálový mechanismus[6] a na základě důkazů získaných výpočetními metodami se zjistilo, že pozorovaná stereospecifita nevylučuje dvoukrokovou adici, při které by vznikal meziprodukt přeměňující se na výslednou látku rychleji než může rotovat, což by umožnilo inverzi stereochemie.
U některých Dielsových–Alderových reakcí dochází při jejich provádění v polárních rozpouštědlech, jako jsou dimethylformamid a ethylenglykol,[12] a dokonce i voda,[13] k výraznému zrychlení průběhu; například reakce cyklopentadienu s butenonem probíhá ve vodě 700krát rychleji než v 2,2,4-trimethylpentanu.[13] Bylo navrženo několik možných vysvětlení tohoto jevu, jako například nárůst efektivní koncentrace kvůli hydrofobnímu uzavírání[14] a stabilizace vodíkové vazby přechodného stavu.[15]
Detaily stereochemie produktu závisí na geometrii dienu a dienofilu. Obzvláště u vnitromolekulárních reakcí je přednostní vznik jednoho z produktů ovlivňován vzájemným působením substituentů reaktantů; významný vliv má ovšem také konformační stabilita struktury přechodného stavu.
Regioselektivita
Teorii předních molekulových orbitalů lze využít i pro vysvětlení regioselektivity pozorované u Dielsových–Alderových reakcí substituovaných reaktantů. Výpočty energií a orbitalových koeficientů předních orbitalů[16] výchozích látek poskytují výsledky, které souhlasí s hodnotami získanými pomocí analýzy rezonančních efektů substituentů:
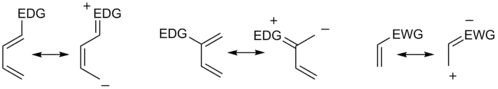
Regioselektivita Dielsových–Alderových reakcí je většinou v souladu s pravidlem ortho-para, které získalo svůj název tak, že vzniklý cyklohexenový produkt má substituenty navázané na pozicích odpovídajících pozicím orto a para u disubstituovaných arenů. Dien se skupinou dodávající elektrony na C1 (prvním uhlíku) má největší HOMO koeficient na C4, zatímco dienofil se skupinou odtahující elektrony na C1 má nejvyšší LUMO koeficient na C2. Spojením vlivu těchto koeficientů dochází k substituci na poloze orto, tak, jak je to níže zobrazeno na obrázku 1. Dien substituovaný na C2, jak je znázorněno na obrázku 2, má nejvyšší HOMO koeficient na C1, a tak se z něj tvoří para produkt. Prozkoumáním uvedeních mezomerních forem ze snadno ověřit, že tyto souhlasí s odhady získanými při uvažování elektronové hustoty a polarizace.
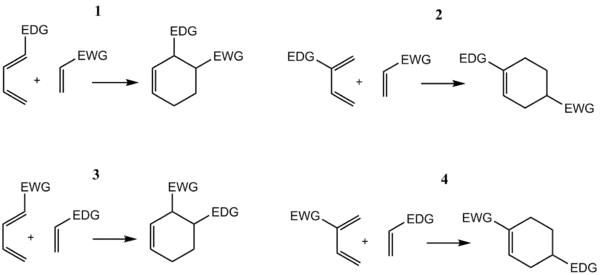
S ohledem na energeticky nejlépe spárované dvojice HOMO-LUMO, tedy na maximalizaci interakční energie při tvorbě vazeb mezi centry s největšími orbitalovými koeficienty, lze odvodit, že regioizomer vznikající v největším množství bude výsledkem takové kombinace dienu s dienofilem.[7] Při pokročileších úvahách se berou v úvahu tři typy substituentů: Z odtahující, tedy snižující HOMO a LUMO koeficienty (CF3, NO2, CN, C(O)CH3), X dodávající, což jsou ty, které zvyšují HOMO a LUMO koeficienty, C spojující, které zvyšují HOMO a snižují LUMO koeficienty (fenyl, vinyl)), což vede k 18 možným kombinacím. Metoda maximalizace orbitalových interakcí správně předpovídá produkt ve všech případech, pro které jsou dostupná experimentální data. Při neobvyklých situacích, jako jsou například přítomnost X dodávajících skupin u dienu i dienofilu, se může přednostně objevovat substituce 1,3.[17] Takové případy jsou však vzácné.
Stereospecifita a stereoselektivita
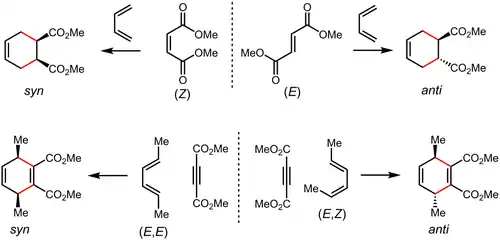
Dielsovy–Alderovy reakce jsou stereospecifické, stereochemie dienu a dienofilu se při nich zachovávají – například pokud jsou substituenty v konfiguraci cis (případně trans), tak dochází ke vzniku produktů, jejichž substituenty jsou vzhledem k cyklohexenovému kruhu ve stejné konfiguraci. Z cis,cis- a trans,trans-disubstituovaných dienů se tvoří produkty s cis substituenty, zatímco při použití cis,trans-disubstituovaného dienu se utvoří sloučenina s trans substituenty.[18][19]
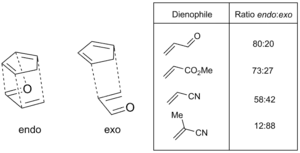
Pokud se na dvou koncích nově vzniklé jednoduché vazby vytvoří stereocentra, pak mohou vznikat dva produkty s odlišnou stereochemií. Jestliže je nejvýznamnější substituent na dienofilu orientován směrem k π systému dienu, pak se vytvoří endo přechodný stav, v opačném případě (nejvýznamnější substituent na dienofilu je orientován směrem od π systému) vzniká exo přechodný stav.
Nachází-li se na dienofilu jeden substituent odtahující elektrony nebo konjugující, případně dva takové substituenty ve vzájemné poloze cis, pak je možné předpovědět, který produkt vznikne. U obvyklých Dielsových–Alderových reakcí je většinou upřednostňován endo meziprodukt, i když je často stericky méně výhodný; tento jev se nazývá Alderovo endo pravidlo; podle Aldera vzniká ve větším množství přechodný stav s „maximálním nahromaděním dvojných vazeb“. Endo selektivita je obvykle výraznější u „tvrdších“ dienofilů jako jsou maleinanhydrid a benzochinon, zatímco při použití jiných, například akrylátů a krotonátů se neobjevuje tak často.[20]
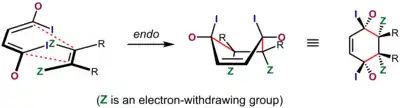
Nejvíce přijímaným vysvětlením tohoto jevu je přednostní interakce mezi π systémy dienofilu a dienu, často nazývané „sekundární orbitalový efekt“, vliv ovšem mohou mít také dipólové a van der Waalsovy síly, selektivita může být též ovlivněna vlastnostmi rozpouštědla.[5][21][22]
Vysvětlení pomocí sekundárního překryvu orbitalů poprvé navrhl Robert Burns Woodward v roce 1917,[23] podle něj se orbitaly spojené při reakci s dvojnou vazbou dienupřekrývají s jeho vnitřními orbitaly, což je možné pouze u endo meziproduktů. I když původní vysvětlení zahrnovalo jen orbital α atomu dvojné vazby dienofilu, tak se následně ukázalo, že se na jevu podílí α i β uhlíkové atomy, pokud to geometrie molekuly umožňuje.[24]
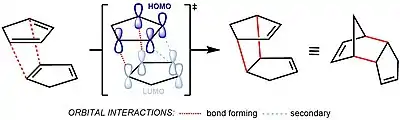
U značně substituovaných dienů či objemných dienofilů mohou sterické efekty převrátit obvyklou endo selektivitu a vést ke vzniku exo izomeru.
Dien
Při Dielsově–Alderově reakci lze použít cyklický i necyklický dien, na kterém navíc mohou být různé substituenty;[5] podmínkou ovšem je, aby se mohl vytvářet s-cis konformaci, jelikož pouze v této konformaci může dojít k požadované reakci.
I když jsou butadieny obvykle stabilnější v konformaci s-trans než v s-cis, tak je rozdíl jejich energií většinou malý (~2–5 kcal/mol).[25]
Dostatečně objemný substituent na C2 nebo C3 může reakci urychlit destabilizací s-trans konformace a vynucením konformace s-cis; například 2-terc-butyl-buta-1,3-dien je 27krát reaktivnější než samotný butadien.[5][26] Dien, který má takové substituenty jak na C2, tak i na C3, bude kvůli sterickým interakcím destabilizujícím konformaci s-cis méně reaktivní.[26]
Dieny s velkými substituenty na koncových uhlících (C1 a C4) rychlost reakce snižují, pravděpodobně kvůli omezení vzájemné přístupnosti dienu a dienofilu.[27]
Obzvlášť reaktivním dienem je 1-methoxy-3-trimethylsiloxy-buta-1,3-dien, známý také jako Danishefského dien,[28] který lze použít v organické syntéze α,β nenasycených systému cyklohexenonů eliminací 1-methoxy substituentu po odstranění enolsilyletherové chránicí skupiny. K významným derivátům 1-methoxy-3-trimethylsiloxy-buta-1,3-dienu patří 1,3-alkoxy-1-trimethylsiloxy-1,3-butadieny (Brassardovy dieny)[29] a 1-dialkylamino-3-trimethylsiloxy-1,3-butadieny (Rawalovy dieny).[30] Zvýšená reaktivita těchto a dalších podobných dienů je způsobena vlivem C1 a C3, které ovlivňují energii HOMO.[3]
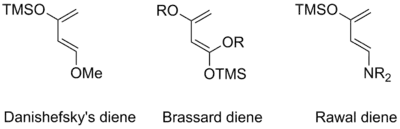
Nestabilní, a tedy vysoce reaktivní, dieny, z nichž se nejčastěji používají o-chinodimethany, lze připravit přímo na místě jejich použití.[31] K běžným metodám přípravy těchto látek patří pyrolýza benzocyklobutenů[5] či odpovídajících sulfonů,[3] 1,4-eliminace orthobenzylsilanů[32] a stannanů[33][34][35] a také redukce α,α'-orthobenzyldibromidů.[36]
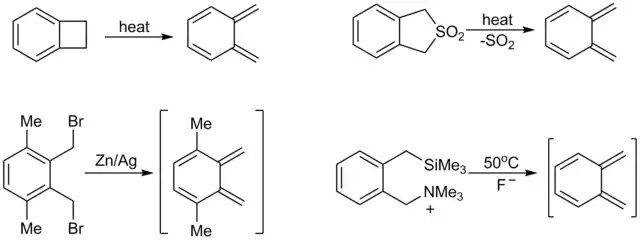
Stabilní dieny jsou často málo reaktivní a Dielsova–Alderova reakce u nich probíhá pouze při vyšších teplotách; jako dien lze použít například naftalen, který však vytváří addukty jen se značně reaktivními dienofily, jako je N-fenyl-maleimid. Antracen, který je méně aromatický (a tedy reaktivnější při Dielsových–Alderových syntézách) může při 80 °C na svém prostředním kruhu vytvořit 9,10 addukt s maleinanhydridem a při 250 °C i s ethynem, který je slabým dienofilem.[37]
Dienofil
U běžné Dielsovy–Alderovy reakce je skupina odtahující elektrony na dienofilu konjugována s alkenem; u inverzní varianty je dienofil naopak konjugován se skupinou. která elektrony přitahuje.[8] V molekule použitého dienu se může nacházet „skrytá funkcionalita“. Dienofil reaguje s dienem za tvorby funkcionality na vzniklé molekule. Požadovaný produkt někdy, pokud je dienofil málo reaktivní nebo obtížně přístupný, nelze získat v jediné reakci; příkladem je použití α-chlorakrylonitrilu (CH2=CClCN). Při reakci s dienem se skrz něj zavede do produktu α-chlornitrilová skupina, která je „skrytou funkcionalitou“, jež může být následně hydrolyzována na keton. α-Chlorakrylonitril je ekvivalentem ketenu (CH2=C=O), jenž vytváří tentýž produkt během jediného kroku. Samotný keten ovšem není možné použít, jelikož reaguje s dieny nežádoucím způsobem ([2+2] cykloadicí) a tak je třeba použít „skrytou funkcionalitu“.[38]
K dalším takto použitelným funkčním skupinám patří fosfoniové substituenty (které po Wittigově reakci dávají vzniknout exocyklickým dvojným vazbám), různé sulfoxidy a sulfonylové sloučeniny (obě jsou ekvivalenty ethenu) a nitroskupiny (ekvivalenty ketenu).[5]
Varianty Dielsovy–Alderovy reakce
Hetero-Dielsova–Alderova reakce
Dielsovy–Alderovy reakce, při kterých alespoň jedna z výchozích látek má v molekule heteroatom, se nazývají hetero-Dielsovy–Alderovy reakce.[39] Například karbonylové sloučeniny mohou reagovat za vzniku dihydropyranových cyklů, k tvorbě dusíkatých heterocyklů lze použít iminy, a to jako dieny i dienofily. Nitrososloučeniny mohou reagovat s dieny za tvorby oxazinů. Chlorsulfonyl-isokyanát je možné použít jako dienofil za účelem přípravy Vinceova laktamu.[5][40]
Aktivace Lewisovou kyselinou
Lewisovy kyseliny jako chlorid zinečnatý, fluorid boritý, chlorid hlinitý a chlorid cíničitý mohou být skrz koordinaci s dienofilem katalyzátory Dielsovy–Alderovy reakce. Vzniklý komplex je lepším elektrofilem a rychleji reaguje s dienem, navíc při tom často dochází ke zlepšení regioselektivity a stereoselektivity. Lewisovy kyseliny rovněž umožňují provádět reakci za nízkých teplot, tedy bez tepelné aktivace.[5]
Asymetrická Dielsova–Alderova reakce
Bylo vyvinuto několik postupů, jak ovlivnit stereoselektivitu Dielsovy–Alderovy reakce; patří k nim použití chirálních pomocníků či nízkomolekulárních organických katalyzátorů[5] a katalýza chirálními Lewisovými kyselinami.[41] Lze také použít Evansovy oxazolidinony,[42] oxazaborolidiny,[43][44][45] cheláty mědi s bis-oxazolinem,[46] a imidazoliny.[47]
Využití
Dielsova–Alderova reakce se využívá při průmyslové výrobě cyklopentadienu, který je prekurzorem řady monomerů. Rovněž nachází využití při výrobě vitaminu B6.
Reference
V tomto článku byl použit překlad textu z článku Diels–Alder reaction na anglické Wikipedii.
- M.C. Kloetzel. The Diels–Alder Reaction with Maleic Anhydride. Organic Reactions. 1948, s. 1–59. ISBN 978-0471264187. (anglicky)
- H.L. Holmes. The Diels-Alder Reaction Ethylenic and Acetylenic Dienophiles. Organic Reactions. 1948, s. 60–173. ISBN 978-0471264187. (anglicky)
- Mehmet Atilla Tasdelen. Diels–Alder "click" reactions: recent applications in polymer and material science. Polymer Chemistry. 2011, s. 2133–2145. Dostupné online. (anglicky)
- G.S. Zweifel; M.H. Nantz. Modern Organic Synthesis: An Introduction. [s.l.]: W.H. Freeman and Co., 2007. ISBN 978-0-7167-7266-8. (anglicky)
- Carey, Part B., pp. 474–526
- M.J. Dewar; S. Olivella; J.J. Stewart. Mechanism of the Diels-Alder reaction: Reactions of butadiene with ethylene and cyanoethylenes. Journal of the American Chemical Society. 1986, s. 5771–5779. PMID 22175326. (anglicky)
- Carey, Part A., pp. 836–50
- Carey, Part A., p. 839
- J.J. Gajewski; K.B. Peterson; J.R. Kagel. Transition-state structure variation in the Diels–Alder reaction from secondary deuterium kinetic isotope effects: The reaction of a nearly symmetrical diene and dienophile is nearly synchronous. Journal of the American Chemical Society. 1987, s. 5545–5546. (anglicky)
- K.N. Houk; Y.T. Lin; F.K. Kagel. Evidence for the concerted mechanism of the Diels–Alder reaction of butadiene with ethylene. Journal of the American Chemical Society. 1986, s. 554–556. PMID 22175504. (anglicky)
- E. Goldstein; B. Beno; K.N. Houk. Density Functional Theory Prediction of the Relative Energies and Isotope Effects for the Concerted and Stepwise Mechanisms of the Diels−Alder Reaction of Butadiene and Ethylene. Journal of the American Chemical Society. 1986, s. 6036–6043. (anglicky)
- R. Breslow; T. Guo. Diels-Alder reactions in nonaqueous polar solvents. Kinetic effects of chaotropic and antichaotropic agents and of β-cyclodextrin. Journal of the American Chemical Society. 1988, s. 5613–5617. (anglicky)
- D.C. Rideout; R. Breslow. Hydrophobic acceleration of Diels-Alder reactions. Journal of the American Chemical Society. 1980, s. 7816–7817. (anglicky)
- R. Breslow; C.J. Rizzo. Chaotropic salt effects in a hydrophobically accelerated Diels–Alder reaction. Journal of the American Chemical Society. 1991, s. 4340–4341. (anglicky)
- Wilfried Blokzijl; Jan B. F. N. Engberts. Initial-State and Transition-State Effects on Diels–Alder Reactions in Water and Mixed Aqueous Solvents. Journal of the American Chemical Society. 1992, s. 5440–5442. (anglicky)
- E.C. Ashby; L.-C. Chao; H.M. Neumann. Organometallic reaction mechanisms. XII. Mechanism of methylmagnesium bromide addition to benzonitrile. Journal of the American Chemical Society. 1973, s. 4896–4904. (anglicky)
- I. Fleming. Frontier Orbital and Organic Chemical Reactions. [s.l.]: Wiley, 1990. ISBN 978-0471018193. (anglicky)
- W. Kirmse; D. Mönch. Umlagerungen von 1,4,4- und 2,2,5-Trimethylbicyclo[3.2.1]oct-6-yl-Kationen. Chemische Berichte. 1991, s. 237–240. (německy)
- G. Bérubé; P. Des Longchamps. Stéréosélection acyclique-1,5: Synthèse de la chaîne latérale optiquement active de la vitamine E. Bulletin de la Société Chimique de France. 1987, s. 103–115. (francouzsky)
- K.N. Houk; L.J. Luskus. Influence of steric interactions on endo stereoselectivity. Journal of the American Chemical Society. 1971, s. 4606–4607. (anglicky)
- Y. Kobuke; T. Sugimoto; J. Furukawa; T. Fueno. Role of attractive interactions in endo–exo stereoselectivities of Diels–Alder reactions. Journal of the American Chemical Society. 1972, s. 3633–3635. (anglicky)
- K.L. Williamson; Y.-F. L. Hsu. Stereochemistry of the Diels–Alder reaction. II. Lewis acid catalysis of syn-anti isomerism. Journal of the American Chemical Society. 1970, s. 7385–7389. (anglicky)
- Robert Burns Woodward; R. Hoffmann. The conservation of orbital symmetry. [s.l.]: [s.n.] ISBN 9781483282046. OCLC 915343522 (anglicky)
- Chaitanya S. Wannere; Ankan Paul; K.N. Houk; Henry F. Schaefer; Paul von Ragué Schleyer. The existence of secondary orbital interactions. Journal of Computational Chemistry. 2007, s. 344–361. ISSN 1096-987X. PMID 17109435. (anglicky)
- Carey, Part A, p. 149
- H.J. Backer. Le 2,3-Ditertiobutylbutadiène. Recueil des Travaux Chimiques des Pays-Bas. 1939, s. 643–661. (francouzsky)
- D. Craig; J.J. Shipman; R.B. Fowler. The Rate of Reaction of Maleic Anhydride with 1,3-Dienes as Related to Diene Conformation. Journal of the American Chemical Society. 1961, s. 2885–2891. (anglicky)
- S. Danishefsky; T. Kitahara. Useful diene for the Diels–Alder reaction. Journal of the American Chemical Society. 1974, s. 7807–7808. (anglicky)
- J. Savard; P. Brassard. Regiospecific syntheses of quinones using vinylketene acetals derived from unsaturated esters. Tetrahedron Letters. 1979, s. 4911–4914. (anglicky)
- S.A. Kozmin; V.H. Rawal. Preparation and Diels−Alder Reactivity of 1-Amino-3-siloxy-1,3-butadienes. Journal of Organic Chemistry. 1997, s. 5252–5253. (anglicky)
- I.L. Klundt. Benzocyclobutene and its derivatives. Chemical Reviews. 1970, s. 471–487. (anglicky)
- Y. Ito; M. Nakatsuka; T. Saegusa. Syntheses of polycyclic ring systems based on the new generation of o-quinodimethanes. Journal of the American Chemical Society. 1982, s. 7609–7622. (anglicky)
- H. Sano; H. Ohtsuka; T. Migita. A convenient method for the generation of o-quinodimethanes by proton induced 1,4-elimination of o-(1-hydroxyalkyl)benzyltributylstannanes. Journal of the American Chemical Society. 1988, s. 2014–2015. (anglicky)
- H.W. Soon. A novel method for the generation of o-quinodimethane by selenium-induced fragmentation of o-vinyl benzyltributylstannane. Tetrahedron Letters. 1993, s. 7587–7590. (anglicky)
- H.W. Soon. Lewis acid-promoted generation of α-oxy-o-quinodimethanes and cycloaddition reactions. Tetrahedron Letters. 1994, s. 3975–3978. (anglicky)
- G. M. Rubottom; J. E. Way. An Improved Method for the Preparation of o-Quinodimethanes. Synthetic Communications. 1984, s. 507–514. (anglicky)
- Margareta Avram (1983). Chimie organica p. 318-323. Editura Academiei Republicii Socialiste România
- S. Ranganathan; D. Ranganathan; A. K. Mehrotra. Ketene Equivalents. Synthesis. 1977, s. 289–296. (anglicky)
- W. R. Roush. Comprehensive Organic Synthesis. Příprava vydání B. M. Trost, I. Flemming. [s.l.]: [s.n.], 1991. ISBN 978-0-08-052349-1. Kapitola Intramolecular Diels–Alder Reactions, s. 513–550. (anglicky)
- P. A. Grieco; S. D. Larsen. Iminium Ion-Based Diels–Alder Reactions: N-Benzyl-2-Azanorborene. Organic Syntheses. 1990, s. 206. Dostupné online. (anglicky)
- James D. White; Subrata Shaw. cis-2,5-Diaminobicyclo[2.2.2]octane, a New Scaffold for Asymmetric Catalysis via Salen−Metal Complexes. Organic Letters. 2011, s. 2488–2491. PMID 21462988. (anglicky)
- D. A. Evans; K. T. Chapman; J. Bisaha. Asymmetric Diels–Alder cycloaddition reactions with chiral α,β-unsaturated N-acyloxazolidinones. Journal of the American Chemical Society. 1988, s. 1238–1256. (anglicky)
- E.J. Corey; T.P. Loh. First application of attractive intramolecular interactions to the design of chiral catalysts for highly enantioselective Diels–Alder reactions. Journal of the American Chemical Society. 1991, s. 8966–8967. (anglicky)
- E.J. Corey; T. Shibata; T. W. Lee. Asymmetric Diels-Alder reactions catalyzed by a triflic acid activated chiral oxazaborolidine. Journal of the American Chemical Society. 2002, s. 3808–3809. PMID 11942799. (anglicky)
- D. H. Ryu; E. J. Corey. Triflimide activation of a chiral oxazaborolidine leads to a more general catalytic system for enantioselective Diels-Alder addition. Journal of the American Chemical Society. 2003, s. 6388–6390. PMID 12785777. (anglicky)
- J. S. Johnson; D. A. Evans. Chiral bis(oxazoline) copper(II) complexes: Versatile catalysts for enantioselective cycloaddition, Aldol, Michael, and carbonyl Ene reactions. Accounts of Chemical Research. 2000, s. 325–335. PMID 10891050. (anglicky)
- K. A. Ahrendt; C. J. Borths; D. W. C. MacMillan. New Strategies for Organic Catalysis: The First Highly Enantioselective Organocatalytic Diels−Alder Reaction. Journal of the American Chemical Society. 2000, s. 4243–4244. (anglicky)
- Arno Behr. Ullmann's Encyclopedia of Industrial Chemistry. [s.l.]: [s.n.], 2000. ISBN 978-3527306732. Kapitola Organometallic Compounds and Homogeneous Catalysis. (anglicky)
Externí odkazy
 Obrázky, zvuky či videa k tématu Dielsova–Alderova reakce na Wikimedia Commons
Obrázky, zvuky či videa k tématu Dielsova–Alderova reakce na Wikimedia Commons