Johnsonova–Coreyova–Čajkovského reakce
Johnsonova–Coreyova–Čajkovského reakce (někdy také nazývaná Coreyova–Čajkovského reakce, zkráceně CCR) je organická chemická reakce používaná na přípravu epoxidů, aziridinů a cyklopropanů. Objevil ji A. William Johnson v roce 1961 a o její výrazné rozvinutí se postarali Elias James Corey a Michael Chaykovsky. Reakce spočívá v adici sirného ylidu na keton, aldehyd, imin nebo enon za vzniku příslušného tříčlenného kruhu. Reakce vykazuje stereoselektivitu ve prospěch trans substituce, a to nezávisle na stereochemii výchozích látek. Tvorba epoxidů tímto způsobem je významnou retrosyntetickou alternativou běžných epoxidačních reakcí alkenů.
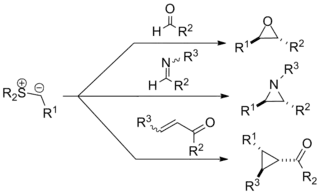
Takto se nejčastěji připravují epoxidy přesunem methylenové skupiny, což bylo využito v několika totálních syntézách (viz oddíl Syntéza epoxidů). Bylo vydáno několik článků zabývajících se touto reakcí.[1][2][3][4][5][6]
Historie
V původním článku popsal A. William Johnson reakce 9-dimethylsulfoniumfluorenylidu se substituovanými deriváty benzaldehydu. Pokus o provedení Wittigovy reakce byl neúspěšný a jako produkt se místo toho vytvořil benzalfluorenoxid.[7]
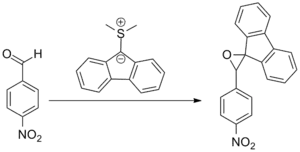
Následný rozvoj (dimethyloxosulfaniumyl)methanidu, (CH3)2SOCH2 a (dimethylsulfaniumyl)methanidu, (CH3)2SCH2 (známých jako Coreyova–Čajkovského činidla) jako činidel zprostředkovávajících přenosy methylenových skupin umožnil zařadit tuto reakci mezi významné součásti organické syntézy.[8]
Mechanismus
Mechanismus Johnsonovy–Coreyovy–Čajkovského reakce obsahuje nukleofilní adici ylidu na karbonylovou nebo iminovou skupinu. Záporný náboj se přesouvá na heteroatom a protože sulfoniový kation patří mezi dobré odstupující skupiny, tak se oddělí a dojde k tvorbě kruhu. U podobné Wittigovy reakce tvorba výrazně silnější dvojné vazby fosfor-kyslík zabraňuje vzniku oxiranu a dochází k alkenaci přes čtyřčlenný cyklický meziprodukt.[4]

Trans diastereoselektivita je způsobena nevratností úvodní adice, která způsobuje, že je rovnováha posunuta směrem k anti betainu oproti syn betainu. Adicí ylidu vznikne betain se sousedícími náboji; na základě výpočtů podle teorie funkcionálu hustoty se ukázalo, že krokem určujícím rychlost reakce je rotace centrální vazby vytvářející konformer potřebný k atakování sulfoniového iontu ze zadní strany.[1]
_V2.png.webp)
Míra vratnosti počátečního kroku (a tedy i diastereoselektivity, která se zvyšuje s rostoucí nevratností) závisí na čtyřech faktorech:[1]
- Stabilitě substrátu: stabilnější substráty vedou k vyšší vratnosti ve prospěch výchozích materiálů oproti betainu.
- Stabilita ylidu: obdobně jako u substrátů i stabilnější ylidy zvyšují vratnost.
- Sterické efekty u betainu ovlivňují vratnost znevýhodněním tvorby meziproduktu a zpomalením rotace centrální vazby, která určuje rychlost reakce.
- Solvatace nábojů betainu protiionty, například Li+: větší míra solvatace ulehčuje rotaci betainu, čímž se vratnost snižuje.
Rozsah
Využití Johnsonovy–Coreyovy–Čajkovského reakce v organické syntéze je široké. Patří pod ní množství reakcí sirných ylidů s elektrofily, i mnoho těch, které nebyly popsány v původních pracích. Je součástí řady totálních syntéz, z nichž jsou některé uvedeny níže, a je v rámci organické chemie považována za silný nástroj.
Druhy ylidů
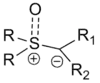
Ylidy mohou obsahovat mnoho různých funkčních skupin, a to jak na aniontovém uhlíku, tak i na atomu síry. Tyto substituenty mohou mít vliv na snadnost přípravy reaktantů (používají se především sulfoniumhalogenidy, jako je trimethylsulfoniumjodid) a rychlost reakce. Na obrázku vpravo je zobrazen obecný vzorec takového ylidu.[1]
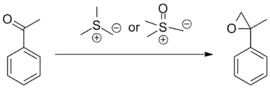
Při použití sulfoxoniových iontů (obsahujících oproti sulfoniovým navíc kyslík vázaný dvojnou vazbou) je příprava činidla snazší a postačují slabší zásady než u sulfoniumylidů. Sulfoniové ionty reagují pomaleji z důvodu vyšší stability. dialkylsulfoxidové vedlejší produkty reakcí sulfoxoniových iontů se navíc vyznačují výrazně nižší toxicitou a těkavostí a slabším zápachem než dialkylsulfidy vznikající ze sulfoniových sloučenin.[1]
Většina používaných reaktantů je monosubstituována na ylidovém uhlíku (kde R1 nebo R2 je atom vodíku). Disubstituované reaktanty jsou také popsány, ovšem používají se méně:[1]
- Reaktanty, které mají na ylidovém uhlíku navázány skupiny odtahující elektrony, se označují jako stabilizované ylidy. Reagují, podobně jako sulfoxoniové sloučeniny, mnohem pomaleji a obvykle se snadněji připravují. Tyto vlastnosti omezují jejich využití: například amidy se používají mnohem častěji než estery a jiné skupiny odtahující elektrony nemají v CCR téměř žádné využití; lépe je lze zapojit do Darzensových reakcí.
- Pokud je na ylidový uhlík navázána arylová nebo allylová skupina, tak se příslušný reaktant řadí mezi semistabilizované ylidy. Tato skupina je široce rozšířená, větší využití mají jen methylenové reaktanty (R1=R2=H). Druh navázané arylové skupiny může značně ovlivnit selektivitu reakce.
- Jestliže je na ylidový uhlík napojena alkylová skupina, pak sloučenina patří k nestabilizovaným ylidům. Na selektivitu má u těchto činidel největší vliv velikost alkylové skupiny.
R skupiny na síře jsou nejběžněji methylové, byly však použity i jiné a podařilo se tak provést i enantioselektivní CCR (viz Varianty). Velikost skupin může měnit také diastereoselektivitu u alicyklických substrátů.[1]
Syntéza epoxidů
Reakcemi sirných ylidů s ketony a aldehydy vznikají epoxidy; tato oblast představuje nejrozšířenější způsob využití Johnsonovy–Coreyovy–Čajkovského reakce. Byly takto použity i složité substráty a „exotické“ ylidy.[9][10]
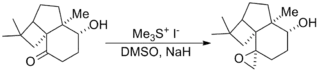
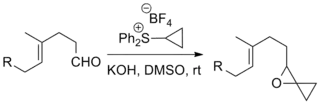
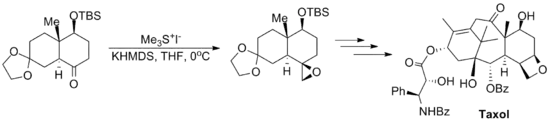
Johnsonovy–Coreyovy–Čajkovského epoxidace byly zahrnuty do několika významných totálních syntéz, například Danishefského totální syntézy taxolu, sloužící k přípravě protinádorového léčiva taxolu, a Kuehneovy totální syntézy strychninu, vytvářející pesticid strychnin.[11][12]

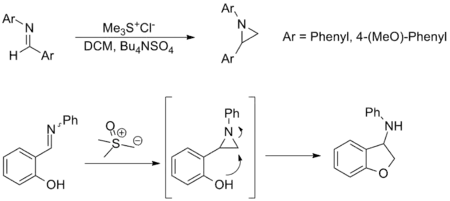
Syntéza aziridinů
Tvorba aziridinů z iminůpatří k dalším významným možnostem použití Johnsonovy–Coreyovy–Čajkovského reakce; představuje alternativu k přesunům aminových skupin z oxaziridinů. Nepoužívá se tak často, vyznačuje se ovšem podobným rozsahem substrátů a funkčních skupin jako karbonylová varianta. Níže jsou zobrazeny příklady; ve druhém z nich se aziridin vytváří in situ a otevírá se nukleofilním atakem, jehož produktem je odpovídající amin.[3][9]
Syntéza cyklopropanů
Při adicích sirných ylidů na enony se se sulfoxoniovými sloučeninami obvykle dosahuje vyšší 1,4-selektivity, než u sulfonových. Lze zde použít mnoho různých skupin odtahujících elektrony, jako jsou ketony, estery a amidy (v příkladu níže je do reakce zapojen Weinrebův amid). U konjugovaných systémů většinou převládají 1,6-adice nad 1,4-adicemi.[3][9]
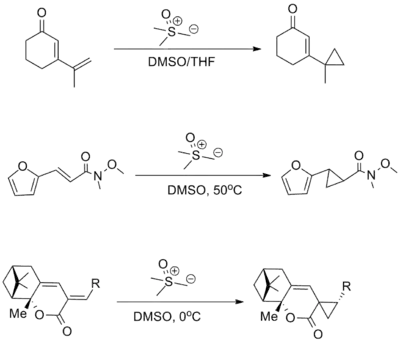
Ostatní reakce
Sirné ylidy lze rovněž použít v řadě podobných homologačních reakcí, pro které se obvykle používá stejné označení.
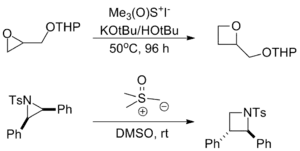
- U epoxidů a aziridinů se takto rozšiřují kruhy za vzniku příslušných oxetanů a azetidinů. Dlouhá reakční doba zabraňuje tomu, aby šlo o významné vedlejší reakce při přípravách epoxidů a aziridinů.[9]
- Jsou známy cykloadice, v nichž ylidy slouží jako nukleofilní ekvivalenty karbenoidů.[9]
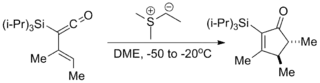
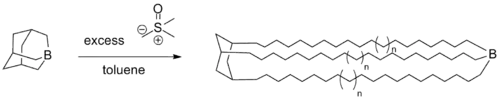
- Také jsou popsány polymerizace s využitím trialkylboranů jako katalyzátorů a (dimethyloxosulfaniumyl)methanidu jako monomeru, použité k syntézám několika komplexních polymerů.[13]
Enantioselektivní varianty
Výzkum enantioselektivních (tedy vytvářejících enantiomerní přebytek) Johnsonových–Coreyových–Čajkovského reakcí neustále probíhá. Použití chirálních sulfidů ve stechiometrických množstvích se ukázalo jako účinnější než odpovídající katalytické postupy, rozsah použitelných substrátů je ale v obou případech omezený. Katalytické varianty byly vyvinuty téměř výhradně pro enantioselektivní varianty; obvyklé organosulfidové reaktanty nejsou příliš nákladné a racemické reakce se dají provést s ekvimolárními množstvími ylidů, aniž by náklady výrazně vzrostly. Chirální sulfidy se obtížně a nákladně připravují, což katalytické enantioselektivní reakce znevýhodňuje.[2]
Stechiometrické reaktanty
Níže jsou zobrazeny neúspěšněji využité stechiometrické reaktanty; prvním je bicyklický oxathian, použitý při syntéze β-adrenergní sloučeniny dichlorisoproterenolu, jeho využití je však omezeno tím, že je dostupný pouze v jednom enantiomeru. Při přípravě axiálního diastereomeru se využívá 1,3-anomeriní efekt, který omezuje nukleofilitu ekvatoriálního volného elektronového páru. Konformace ylidu je omezená Prelogovým napětím a v důsledku sterických interakcí s methylovými substituenty se aldehyd k ylidu může přiblížit pouze z jedné strany.[5][2]
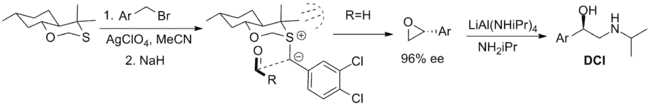
Další významný reaktant, odvozený od kafru, vyvinul Varinder Aggarwal. Oba enantiomery této sloučeniny se získávají snadno, i když výtěžky jsou menší než u oxathianu. Konformaci ylidu určují interakce s vodíkovými atomy na můstku spojujícím cykly a přístupu aldehydu zamezuje kafrová funkční skupina. Při reakci se používá fosfazen jako zásada, která vyvolává tvorbu ylidu.[5][2]

Katalytické reaktanty
Katalytické reaktanty byly použity s menším úspěchem, většina reakcí měla nízkou výtěžnost a nebo špatnou enantioselektivitu. Také spektrum substrátů bylo omezené, obzvláště u přenosu methylenu, hlavně v rámci alifatických aldehydů. Bylo potřeba najít nukleofilní sulfid, který vytváří ylid s velkou účinností a současně je dobrou odstupující skupinou (pro tvorbu epoxidu). Protože jsou tyto vlastnosti v protikladu, tak se nalezení vhodného katalyzátoru ukázalo jako obtížné. Níže je zobrazeno několik nejúčinnějších katalyzátorů a výtěžnosti a enantiomerní přebytky při přípravě (E)-stilbenoxidu.[5][2]
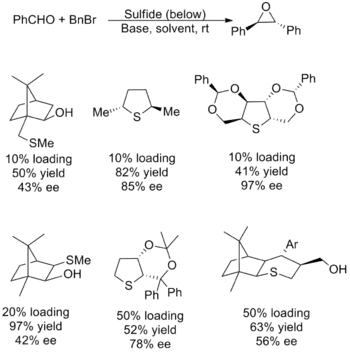
Byl také vyvinut postup založený na stejném sulfidu a zahrnující také alkylaci pomocí in situ vytvořeného rhodiového karbenoidového katalyzátoru. Tato metoda měla také úzký rozsah substrátů, kdy neprobíhala s žádným elektrofilem majícím zásadité substituenty, protože se v takových případech objevovaly vedlejší reakce spotřebovávající karbenoidy.[2]

Odkazy
Reference
V tomto článku byl použit překlad textu z článku Johnson–Corey–Chaykovsky reaction na anglické Wikipedii.
- V. K. Aggarwal; J. Richardson. The complexity of catalysis: origins of enantio- and diastereocontrol in sulfur ylide mediated epoxidation reactions. Chemical Communications. 2003, s. 2644–2651. DOI 10.1039/b304625g. PMID 14649793.
- V. K. Aggarwal; C. L. Winn. Catalytic, Asymmetric Sulfur Ylide-Mediated Epoxidation of Carbonyl Compounds: Scope, Selectivity, and Applications in Synthesis. Accounts of Chemical Research. 2004, s. 611–620. DOI 10.1021/ar030045f. PMID 15311960.
- Y. G. Gololobov; A. N. Nesmeyanov; V. P. Lysenko; I. E. Boldeskul. Twenty-five years of dimethylsulfoxonium ethylide (corey's reagent). Tetrahedron. 1987, s. 2609–2651. DOI 10.1016/s0040-4020(01)86869-1.
- A.-H. Li; L.-X. Dai; V. K. Aggarwal. Asymmetric Ylide Reactions: Epoxidation, Cyclopropanation, Aziridination, Olefination, and Rearrangement. Chemical Reviews. 1997, s. 2341–2372. DOI 10.1021/cr960411r. PMID 11848902.
- V. K. Aggarwal; J. Gair Ford; Sílvia Fonguerna; Harry Adams; Ray V. H. Jones; Robin Fieldhouse. Catalytic Asymmetric Epoxidation of Aldehydes. Optimization, Mechanism, and Discovery of Stereoelectronic Control Involving a Combination of Anomeric and Cieplak Effects in Sulfur Ylide Epoxidations with Chiral 1,3-Oxathianes. Journal of the American Chemical Society. 1998-08-08, s. 8328–8339. DOI 10.1021/ja9812150.
- E. M. McGarrigle; E. L. Myers; O. Illa; M. A. Shaw; S. L. Riches; V. K. Aggarwal. Chalcogenides as Organocatalysts. Chemical Reviews. 2007, s. 5841–5883. DOI 10.1021/cr068402y. PMID 18072810.
- A. W. Johnson; R. B. LaCount. The Chemistry of Ylids. VI. Dimethylsulfonium Fluorenylide—A Synthesis of Epoxides. Journal of the American Chemical Society. 1961, s. 417–423. DOI 10.1021/ja01463a040.
- E. J. Corey; M. Chaykovsky. Dimethyloxosulfonium Methylide ((CH3)2SOCH2) and Dimethylsulfonium Methylide ((CH3)2SCH2). Formation and Application to Organic Synthesis. Journal of the American Chemical Society. 1965, s. 1353–1364. DOI 10.1021/ja01084a034.
- Jack Jie Li. Named Reactions in Heterocyclic Chemistry. [s.l.]: John Wiley & Sons, 2005. Dostupné online. ISBN 9780471704140. S. 2–14.
- P. Mundy Bradford; Michael D. Ellerd; Frank G. Favaloro. Name Reactions and Reagents in Organic Chemistry. [s.l.]: John Wiley & Sons, 2005. Dostupné online. ISBN 9780471739869. S. 174–175, 743.
- S. J. Danishefsky, J. J. Masters, W. B. Young, J. T. Link, L. B. Snyder, T. V. Magee, D. K. Jung, R. C. A. Isaacs, W. G. Bornmann, C. A. Alaimo, C. A. Coburn, M. J. Di Grandi. Total Synthesis of Baccatin III and Taxol. Journal of the American Chemical Society. 1996, s. 2843–2859. DOI 10.1021/ja952692a.
- M. E. Kuehne; F. Xu. Total synthesis of strychnan and aspidospermatan alkaloids. 3. The total synthesis of (.+-.)-strychnine. The Journal of Organic Chemistry. 1993, s. 7490–7497. DOI 10.1021/jo00078a030.
- J. Luo; K. J. Shea. Polyhomologation. A Living C1 Polymerization. Accounts of Chemical Research. 2010, s. 1420–1433. DOI 10.1021/ar100062a. PMID 20825177.
Související články
- Darzensova reakce
- Wittigova reakce
- Epoxidace
- Ylidy
- Totální syntéza taxolu
- Totální syntéza strychninu
Externí odkazy
 Obrázky, zvuky či videa k tématu Johnsonova–Coreyova–Čajkovského reakce na Wikimedia Commons
Obrázky, zvuky či videa k tématu Johnsonova–Coreyova–Čajkovského reakce na Wikimedia Commons - Animace mechanismu
{kind=link}