Organická syntéza
Organická syntéza je specificky zaměřený obor chemické syntézy, který se zabývá stavbou organických sloučenin za pomoci organických reakcí. Organické molekuly jsou mnohdy složitější ve srovnání s čistě anorganickými sloučeninami, takže se syntéza organických sloučenin rozvinula v jedno z nejdůležitějších odvětví organické chemie. Dělení typů organických syntéz je velice ošidné z důvodu neustálého bouřlivého vývoje, a proto jsou zde uvedeny pouze základní reakce používané při organických syntézách.
Redukce
Úvod
Transformace organických sloučenin redukcí či oxidací patří mezi běžné a široce rozvinuté operace v organické syntéze. V anorganické chemii je redukce/oxidace definována jako přijímání/ztráta elektronů. To u organické chemie nejde, tudíž se při pozorování oxidačního stavu organické molekuly v organické chemii při redoxních reakcích používají určité účelové úpravy (zde jsou uvedeny jen ty nejvýznamnější):
- Atomy uhlíku v uhlíkatém řetězci vystupují navzájem s oxidačním číslem 0.
- Každá skupina s jedním atomem uhlíku je navenek bez náboje (elektroneutrální).
- Atom vodíku má vždy formální oxidační číslo +I.
- Atom kyslíku má s výjimkou peroxidů formální oxidační číslo -II.
Tudíž např. v molekulách ethanolu a ethanalu budou oxidační čísla následující:
-III . -I
CH3-CH2-OH
-III . +I
CH3-CH=O
Protože redoxní reakce jsou vždy reakcemi komplementárními, v níž se jedna sloučenina redukuje/oxiduje a druhá oxiduje/redukuje, posuzujeme v organické chemii tyto procesy z hlediska změny oxidačního čísla organické látky a sledujeme pouze změny, kterým podléhají konkrétní atomy, skupiny atomů a funkční skupiny.
Redukce: CH3-CH=O → CH3-CH2-OH (+I → -I)
Oxidace: CH3-CH2-OH → CH3-CH=O (-I → +I)
Reakce
Z mechanického hlediska se uvádí tři hlavní kategorie:
- 1) Připojení vodíku k násobné vazbě
K těmto reakcím řadíme např.: Katalytickou hydrogenaci alkenů, alkynů a funkčních skupin s násobnými vazbami či katalytickou hydrogenolýzu vazeb.
R-CH=CH2 + H2 → R-CH2-CH3
Ar-CH2-N(CH3)2 + H2 → Ar-CH3 + (CH3)2NH
2) Adice elektronů, obvykle následována připojením protonu.
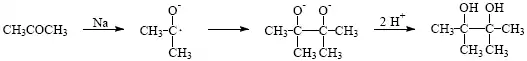
3) Adice hydridového iontu, obvykle z komplexního hydridu, na polární násobné vazby např. C=O, COOR, C=N

Oxidace
Úvod
Oxidativní přeměny jsou po redukci jednou z nejdůležitějších metod změny oxidačního stavu funkčních skupin substrátu. Za oxidaci je považován každý proces, ve kterém sloučenina nebo skupina odevzdává elektrony. V organické syntéze se však většinou omezujeme na souhrn transformací při kterých dochází k jedné z následujících oxidativních přeměn organického substrátu:
- 1) Přenos elektronu z organické molekuly na činidlo
![]()
- 2) Přijímání atomu kyslíku na atom uhlíku nebo heteroatom vázaný v molekule
RCHO + O → RCOOH
- 3) Snižování počtu atomů vodíku vázaných na atomu uhlíku nebo heteroatomu.
RCH2OH → RCHO + 2H
Reakce
Oxidativní přeměny probíhají řadou rozdílných mechanismů.
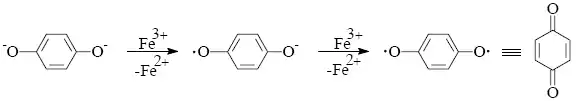
1) Jednoelektronové oxidace činidlem s vhodným redoxním potenciálem schopným jednoelektronové redukce (např. Fe3+ → Fe2+) probíhají úspěšně tehdy, vzniká-li oxidací relativně stálý radikál. Tímto mechanismem probíhá např. oxidace dvousytných fenolů na chinony.
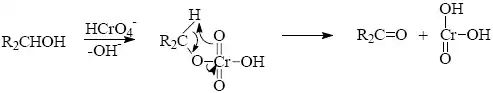
2) Dvouelektronová oxidace iontem kovu schopným dvouelektronové redukce obvykle probíhá přes stádium intermediátu, např. oxidace alkoholu na karbonylové sloučeniny.
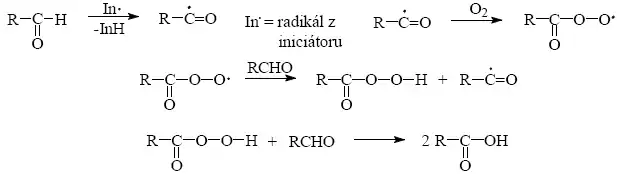
3) Odstraňování atomu vodíku na aktivovaném místě molekuly např. radikálová řetězová autooxidace aldehydů.
- 4) Další reakce
Přenos hydridového iontu na jinou molekulu (Cannizarrova reakce), připojení kyslíku do organické molekuly (Bayer-Villigerova oxidace), aj.
Halogenace
Úvod
Halogenací obecně označujeme nejen reakce, kdy halogen přímo působí na organický substrát, ale také všechny reakce, při kterých se vytváří vazba uhlík-halogen. Halogenderiváty hrají hlavní úlohu v alkylacích a arylacích při zavádění uhlíkatých řetězců do organické molekuly, neboť halogenové ionty patří mezi dobře odstupující částice. Z těchto důvodů obvykle halogenderiváty nepředstavují koncové produkty, ale reaktivní intermediáty v dalších transformacích.
Alifatické halogenderiváty
Pro zavedení halogenu do alifatického řetězce se využívá rozdílných substrátů a reakcí probíhající rozdílnými mechanismy, např. homolytickou substitucí alkenů a alkynů, nukleofilní substitucí kyslíkaté skupiny v alkoholech, etherech a karboxylových kyselinách, aj.
Reakce
- 1) Halogenace adičními reakcemi
Adice halogenovodíků HX (X=F,Cl,Br,I) na alkeny a alkyny byla široce studována jak z mechanického hlediska, tak z praktického hlediska. Její význam je však limitován nežádoucími vedlejšími reakcemi. Snadnost adice halogenovodíků na jednoduché alkeny je v souladu s jejich klesající aciditou HI > HBr > HCl > HF. Adice obvykle probíhají regioselektivně a řídí se polarizací násobné vazby.
Stereoselektivita reakce závisí na mechanismu. Rozlišují se dva případy: via karbokation vznikající protonizací násobné vazby či termonukleární mechanismus spočívající v nukleofilním ataku na komplex alken-HX.
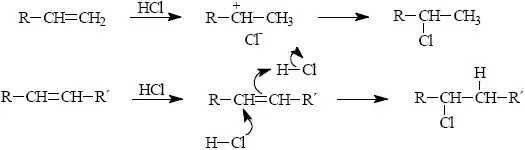
Rozhodujícím faktorem určujícím typ mechanismu je stabilita intermediárního karbokationtu. Alkeny, které snadno vytvářejí stálý karbokation, preferují spíše mechanismus přes iontový pár a tedy cis-adici. termonukleární mechanismus preferuje trans-připojení na násobnou vazbu.
Dále můžeme zmínit adici halogenů, interhalogenů, hypohalogenkyselin a příbuzných činidel.
- 2) Substituční halogenace
Substituce vodíku halogenem.Alkany podléhají radikálové substituci fluorem, chlorem a bromem. Reakce s fluorem je nekatalyzovaná, substituci ostatních halogenů je nutno iniciovat vhodným způsobem (teplo, UV-záření, aj.)
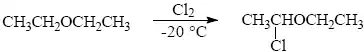
Substituce kyslíkatých funkčních skupin. Nejběžnějšími edukty halogenderivátů jsou odpovídající alkoholy. Protože hydroxyskupina je v nukleofilních substitucích skupinou špatně odstupující, musí se nejdříve aktivovat.
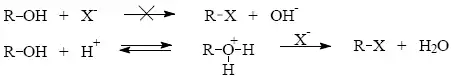
Substituce halogenů. Náhradou halogenu za jiný typ halogenu obvykle sledujeme změnu reaktivity halogenderivátu nebo změnu jeho užitných vlastností.
![]()
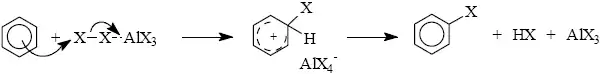
Aromatické halogenderiváty
Zavádění halogenů na aromatické jádro elektrofilní substitucí je důležitým syntetickým pochodem. Chlor a brom jsou vůči aromatickým uhlovodíkům aktivní. Fluorace elementárním fluorem je silně exotermická, obtížně kontrolovatelná a zpravidla se volí činidla modifikovaná. Jodace elementárním jódem probíhá jen u reaktivních substrátů a provádí se obvykle aktivnějšími činidly. Mezi nejmírnější činidla pro chloraci a bromaci patří elementární halogeny, obvykle rozpuštěné v kyselině octové nebo v halogenovém rozpouštědle jako tetrachlormethan nebo chloroform.
Nitrace, nitrosace a sulfonace
Při přípravě nitro- a nitrosolátek, resp. sulfonových kyselin využívá organická syntéza řadu postupů. Zde je zatím uvedena pouze substituce vodíkového atomu za nitro-, nitroso- či sulfoskupinu (za adice na dvojné vazbě).
Nitrace
- 1) Nitrace alifatických sloučenin
Přímá nitrace alkanů kyselinou dusičnou nebo oxidy dusíku (N2O4,N2O5) má využití pouze v průmyslové sféře. V laboratoři je význam těchto metod značně omezen na jednoduché alkany a cykloalkany, přesto nitrací vznikají obvykle směsi sloučenin. Alifatické nitrosloučeniny vznikají např. adicí halogenidu nitrilu na dvojnou vazbu.
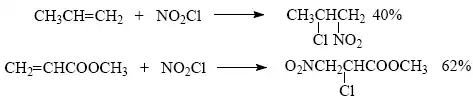
- 2) Nitrace aromatických sloučenin
Elektrofilní aromatická nitrace je důležitá metoda nejen pro tvorbu aromatických dusíkatých sloučenin. Transformací nitroskupiny a následnými přeměnami lze do aromatického jádra zavést řadu funkčních skupin. Aromatické nitraci podléhají i velmi málo reaktivní substráty, neboť dnes je k dispozici široká nabídka nitračních činidel. Nitrací fenolů a fenoletherů (již zředěnou kyselinou dusičnou) lze připravit monoderiváty.
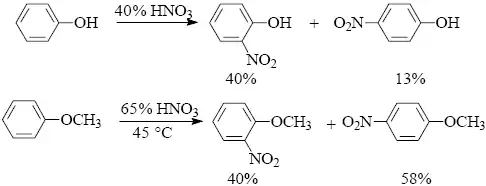
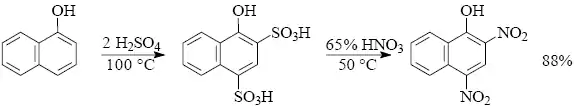
Při nitraci fenolů do vyšších stupňů je třeba eliminovat nežádoucí oxidační působení nitrační směsi. Fenol se proto nejprve sulfonuje koncentrovanou kyselinou sírovou a pak teprve následuje nitrace.
Nitrosace
- 1) Nitrosace alifatických sloučenin
Radikálová nitrosace alkanů není preparativně rozšířená, realizuje se v průmyslovém měřítku působením chloridu nitrosylu za iniciace ultrafialovým zářením.
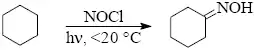
- 2) Nitrosace aromatických sloučenin
Na rozdíl od nitrace podléhají elektrofilní nitrosaci jen aktivované substráty, tj. aminy a fenoly. Nejběžnějšími nitrosačními činidly jsou kyselina dusitá uvolňovaná obvykle z alkalického dusitanu a minerální kyseliny. Omezeně byl pro nitrosace používán i chlorid nitrosylu a oxid dusitý. Nitrosace fenolů probíhá přednostně do p-polohy, pouze v případě, kdy je tato poloha obsazena, vstupuje nitrososkupina do polohy ortho.
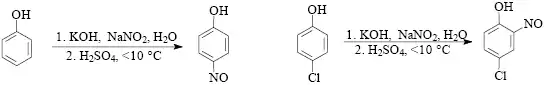
Sulfonace
Sulfonací se v obvyklém slova smyslu rozumí reakce, při níž se substitucí atomu vodíku v molekule substrátu zavádí sulfoskupina SO3H nebo skupiny od ní odvozené, přitom vzniká vazba C-S.
- 1) Sulfonace alifatických sloučenin
Alifatické sloučeniny podléhají sulfonaci oxidem sírovým nebo činidly od něj odvozenými jen zřídka. Běžně se pro přípravu sulfonových kyselin využívají radikálové reakce s účastí oxidačního činidla (sulfochlorace, sulfooxidace) nebo reakce alkylační.
![]()
- 2) Sulfonace aromatických sloučenin
Sulfonace aromatických sloučenin je mnohostranně využitelná reakce a podléhá jí většina aromatických systémů včetně kondenzovaných. Na reaktivitě substrátu - analogicky jako při nitraci - závisí volba činidla. Sulfonace je z elektrofilních aromatických substitucí výjimečná v tom, že je vratná a sulfoskupinu je možno z molekuly aromatické sloučeniny opět odstranit. Dalším fenoménem sulfonace je možnost ovlivnit distribuci produktů kinetickým nebo termodynamickým řízením reakce. Tuto skutečnost lze dokumentovat na příkladu sulfonace fenolu koncentrovanou kyselinou sírovou.
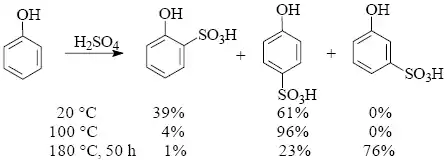
Alkylace a acylace
Alkylací a acylací nazýváme reakce, při nichž dochází k připojení alkylu nebo acylu k atomu uhlíku nebo heteroatomu (P,S,O,N,atd..). Za alkylační reakce lze považovat také přípravu halogenderivátu nebo alkylaci kovů.
Alkylace alifatických sloučenin
V alifatických substrátech probíhají reakce obecně tímto způsobem:
![]()
Alkylační činidlo jakožto elektrofil reaguje s nukleofilem, který má k dispozici volný elektronový pár. Mechanismus reakce závisí především na povaze alkylačního činidla, charakteru nukleofilu a reakčních podmínkách. Síla alkylačního činidla se určuje podle síly kyseliny, od které je činidlo odvozeno. Mezi silná alkylační činidla patří např. dialkyl-sulfáty, naopak mezi slabé spadají např. halogenderiváty nebo estery arensulfonových kyselin.
Nukleofilem může být každá částice s nesdíleným elektronovým párem.
Příprava a reakce organokovových sloučenin
Jako organokovové sloučeniny nazýváme látky, v nichž je atom kovu M vázán na atom uhlíku. Vlastnosti vazby mezi uhlíkem a kovem závisí především na vlastnostech kovu, rozpouštědle a také na struktuře uhlíkatého zbytku.
Náhrada funkční skupiny kovem
Nejdůležitější metoda přípravy organokovových sloučenin je Reakce halogenderivátů s kovy. Nejpoužívanější k těmto reakcím jsou především skupiny halogenidů.
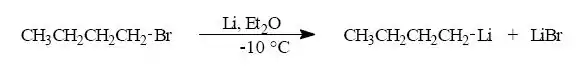
Náhrada vodíku kovem
Reakci, při níž dojde k náhradě vodíku za kov, umožňují pouze velmi silné C-kyseliny. Význam reakcí je však pouze omezený. Mezi nejběžnější patří např. tvorba organolithných sloučenin.
Výměna kovu za kov
Výměna kovu za kov (tzv. transmetalace) je velmi často používanou a univerzální metodou přípravy organokovových sloučenin. Nejjednodušším způsobem, jak dosáhnout transmetalace je přímá substituce organokovové sloučeniny kovem jiným. Toho je možné dosáhnout pouze v případě, že kov, kterým chceme substituovat je elektropozitivnější, než kov vázaný ve sloučenině. Tímto postupem je možno dosáhnout velmi čistých organokovů bez příměsí jakýchkoliv solí.
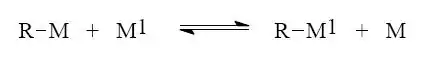
Druhá metoda zahrnuje výměnu kovu mezi organokovovým činidlem (nejčastěji Grignardovým) a halogenidem kovu.

Velmi ojedinělou metodu představuje výměnná reakce mezi dvěma organokovovými činidly. Tato metoda se využívá především k přípravě organolithných činidel.
Příprava a reakce diazoniových solí
Tvorba diazoniových solí
Diazoniové soli vznikají pouze v kyselém prostředí při reakci primárních aminů s kyselinou dusitou. Princip diazotace je v reakci elektrofilního nitrosoniového iontu s nukleofilním dusíkem aminu.
Diazotaci podléhají jak alifatické, tak aromatické aminy. První jmenované jsou však velmi nestabilní a rozpadají se na molekulární dusík a reaktivní karbokation. Aromatické diazoniové ionty jsou za nízkých teplot relativně stabilní. Za určitých podmínek mohou být diazoniové ionty izolovány jako soli (např. BF4-; CF3COO-, atd.)
Reakce diazoniových solí
- Monomolekulární rozklad diazoniového iontu
- Při této reakci vzniká velmi nestabilní fenylový kation, díky tomu se ke iontu může připojit jakýkoliv nukleofil za vzniku neutrální molekuly.
- Jednoelektronový přenos
- Tento mechanismus se uplatňuje v reakcích, kde jako katalyzátor vystupuje měď nebo její soli.
Eliminace
Rozlišujeme 2 základní typy eliminace.
1. α-eliminace
Při tomto druhu eliminace se dva atomy nebo skupiny atomů oddělí z jednoho a téhož atomu. Výsledkem je nestálá částice.
2. β-eliminace V tomto případě se atomy nebo skupiny atomů oddělí ze dvou sousedních atomů za vzniku násobné vazby.
a. Monomolekulární reakce
b. Bimolekulární reakce
Dehydratace
Dehydratace alkoholů
Dehydratací alkoholů rozumíme eliminaci vody z alkoholů účinkem kyselých katalyzátorů, nejčastěji se provádí pomocí kyseliny sírové nebo fosforečné. Nejčastěji se dehydratují terciární alkoholy, dehydratace sekundárních a primárních alkoholů vyžaduje koncentrovanou kyselinu a vysoké teploty.
Extruze
Extruze jsou tepelně nebo fotochemicky vyvolané reakce, při nichž atom, nebo skupina atomů Y vázaná k jiným atomům X, Z, se eliminuje z molekuly substrátu za vzniku nového produktu X-Z.

Extruze se využívá především k výrobě alkenů a cyklických sloučenin.
Externí odkazy
 Obrázky, zvuky či videa k tématu Organická syntéza na Wikimedia Commons
Obrázky, zvuky či videa k tématu Organická syntéza na Wikimedia Commons
Literatura
- SVOBODA, J. Organická syntéza I. Praha: Vysoká škola chemicko-technologická v Praze, 2000. Dostupné online. ISBN 80-7080-385-1. S. 302. (česky)
- DĚDEK, Václav; ČERVINKA, Otakar; FERLES, Miloslav. Organická chemie. Bratislava: SNTL / ALFA, 1980. 04-609-80. S. 792. (česky)