Organolithné sloučeniny
Organolithné sloučeniny jsou skupinou organokovových sloučenin, které mají v molekule jeden či více atomů lithia vázaných na uhlíkové atomy. Jsou významnými reaktanty v organické syntéze; často se používají k přenosu organické skupiny nebo atomu lithia na substráty deprotonací nebo nukleofilní adicí.[1] V průmyslu se používají na zahájení aniontové adiční polymerizace, při níž se vyrábějí různé elastomery. Organolithné sloučeniny rovněž nacházejí využití při asymetrické syntéze při výrobě léčiv.
Vzhledem k velkému rozdílu elektronegativity mezi atomy uhlíku a lithia má vazba C-Li iontový charakter. Díky tomu jsou organolithná činidla dobrými nukleofily a silnými zásadami. Pro laboratorní použití je komerčně dostupných mnoho takovýchto látek ve formě roztoků.
Organolithné sloučeniny jsou velmi reaktivní a někdy i samozápalné.

Historie a vývoj
Organolithné sloučeniny začali v roce 1930 studovat Karl Ziegler, Georg Wittig a Henry Gilman. Zjistili, že použití organolithných sloučenin často vede k vyšším rychlostem i výtěžnostem reakcí než použití Grignardových činidel.[2] Od této doby organolithné sloučeniny v mnoha případech nahradily Grignardova činidla. Následující výzkum byl zaměřen na povahu vazby uhlík-lithium, strukturní studium organolithných komplexů, chirální organolithná činidla a asymetrickou syntézu.[3]
Struktura
I když se jednoduché alkyllithné sloučeniny obvykle popisují jako monomery RLi, tak existují jako shluky (oligomery) nebo polymery.[4] Míra shlukování záleží na organickém substituentu a na přítomnosti dalších ligandů.[5][6] Tyto struktury byly zjištěny mnoha metodami, mezi něž patří 6Li, 7Li a 13C NMR spektroskopie a rentgenová difrakční analýza[1] a následně potvrzeny metodami výpočetní chemie.[4]
Vazba uhlík-lithium
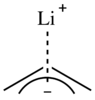
Na základě vysokého rozdílu elektronegativity mezi uhlíkem a lithiem lze očekávat značnou polaritu této vazby;[7][8][9] některé organolithné sloučeniny ovšem vykazují vlastnosti nepolárních látek jako je například rozpustnost v nepolárních rozpouštědlech.[7] I když z většiny dat vyplývá iontová povaha vazby C-Li, uvažuje se o tom, že by mohla mít slabě kovalentní charakter.[8][9]
Struktura v pevném skupenství
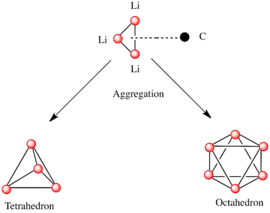
Organolithné sloučeniny, podobně jako jiné látky s polárními podjednotkami, vytvářejí shluky.[6][10] Tvorba těchto shluků je ovlivňována elektrostatickými interakcemi, koordinací mezi lithiem a okolními molekulami rozpouštědla a sterickými efekty.[6]
Základní stavební jednotkou při utváření složitějších struktur představuje karbaniontové centrum interagující s Li3 trojúhelníkem.[4]
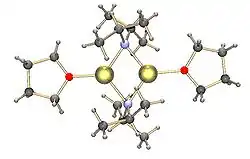
Běžné amidy lithia, například bis(trimethylsilyl)amid lithný a diisopropylamid lithný, také podléhají shlukování.[11] Amidy lithia zaujímají v nekoordinačních rozpouštědlech v pevném skupenství polymerní žebříkovitou strukturu a v etherových rozpouštědlech se vyskytují jako dimery. Za přítomnosti silně donujících ligandů se vytvářejí tri- nebo tetramerní lithiová centra.[12]; například LDA ( diisopropylamid lithný) se v THF vyskytuje převážně v dimerní formě.[11] Struktura běžnějších amidů lithia, například LDA a hexamethyldisilazidu lithného (LiHMDS) byla podrobně zkoumána pomocí NMR spektroskopie.[13] Další významnou skupinou organolithných sloučenin jsou silyllithné sloučeniny, často využívané při syntézách organokovových komplexů a polysilanových dendrimerů.[6][14] Na rozdíl od alkyllithných sloučenin většina silyllithných činidel v pevném skupenství monomerní struktury koordinované s molekulami rozpouštědla, například THF, a jen u několika málo silyllithných sloučenin byly popsány vícenásobné shluky.[6] Tento rozdíl může být způsoben způsobem přípravy, sterickými jevy způsobenými velkými alkylovými substituenty na křemíkovém atomu, a také menší polaritou vazby Si-Li. Koordinované molekuly rozpouštědla lze odstranit přidáním silně donujících činidel, jako jsou TMEDA a (−)-spartein.[6]
Struktura v roztocích
Při určování struktury v roztocích nelze zcela spoléhat na strukturní informace o organolithných shlucích v pevném skupenství získané u jejich krystalové struktury, jelikož mohou tyto látky v roztocích zaujímat odlišné struktury.[5] V některých případech může být navíc obtížné získat je v krystalické podobě. Studium struktury roztoků organolithných činidel a meziproduktů obsahujících lithium je však značně užitečné k pochopení jejich reaktivity.[15] Velmi užitečnou metodou pro studium struktury organolithných shluků v roztocích je NMR spektroskopie. U alkyllithných sloučenin lze k určení počtu atomů lithia interagujících s karbaniontovými centry, a zda jsou tyto interakce statické či dynamické, použít C-Li J párování.[5] Separate NMR signals can also differentiate the presence of multiple aggregates from a common monomeric unit./překlad[16]
Strukturu organolithných sloučenin může ovlivnit přítomnost Lewisovy zásady jako například tetrahydrofuranu (THF), diethyletheru (Et2O), tetramethylethylendiaminu (TMEDA) nebo hexamethylfosforamidu (HMPA).[4] Zvláštním případem je methyllithium, u něhož při rozpuštění v diethyletheru nebo HMPA nedochází k zániku tetramerní struktury.[6] THF deagreguje hexamertní butyllithium, poté převažuje tetramer; ΔG přeměny tetrameru na dimer je přibližně 46 kJ/mol.[17] TMDA také způsobuje chelaci lithných kationtů v n-butyllithiu a vytváří solvatované dimery jako je [(TMEDA) LiBu-n)]2.[4][5] U fenyllithia bylo zjištěno, že se v krystalizované solvatované etherové formě vyskytuje jako zkřivený tetramer a v etherovém roztoku jako směs dimeru a tetrameru.[5]
| Rozpouštědlo | Struktura | |
|---|---|---|
| methyllithium | THF | tetramer |
| methyllithium | diethylether/HMPA | tetramer |
| n-butyllithium | pentan | hexamer |
| n-butyllithium | diethylether | tetramer |
| n-butyllithium | THF | tetramer-dimer |
| s-butyllithium | pentan | hexamer-tetramer |
| isopropyllithium | pentan | hexamer-tetramer |
| t-butyllithium | pentan | tetramer |
| t-butyllithium | THF | monomer |
| fenyllithium | ether | tetramer-dimer |
| fenyllithium | diethylether/HMPA | dimer |
Struktura a reaktivita
Struktura, a tedy i jejich reaktivita, se mění v závislosti na chemickém prostředí.[6][18] Jedním z problému je to, jestli existuje nějaký vztah mezi mírou shlukování a reaktivitou organolithných sloučenin. Původně se předpokládalo, že jednodušší shluky (jako jsou monomery) jsou reaktivnější u alkyllithných sloučenin.[19] Byly však objeveny také reakce, u kterých je reaktivní látkou také dimer nebo jiný oligomer[20] a u lithných amidů (například LDA) jsou běžné reakce založené na dimerech.[21] Několik studií kinetiky reakcí v roztocích řízených LDA ukázalo, že jednodušší shluky enolátů nemusejí nutně mít vyšší reaktivitu.[13]
Reaktivitu organolithných sloučenin zvyšují rovněž některé Lewisovy zásady;[22] [23] není ovšem známo, zda tyto látky nefundují jako silná chelatační činidla a jak pozorované zvýšení reaktivity závisí na jimi způsobených změnách struktury.[22][23] Jako příklad lze uvést TMEDA, jenž zvyšuje rychlost i výtěžnost mnoha reakcí organolithných sloučenin.[6] Vzhledem k alkyllithným sloučeninám funguje TMEDA jako donorový ligand, snižuje úroveň shlukování,[4] a zvyšuje nukleofilitu těchto látek.[24] TMEDA ovšem nefunguje vždy jako donorový ligand lithného kationtu, obzvláště za přítomnosti aniontových kyslíkových a dusíkových center, například s LDA a LiHMDS interaguje jen slabě i v uhlovodíkových rozpouštědlech bez „soutěžících“ donorových ligandů.[25] U lithiace iminů, kde je THF silným donorovým ligandem pro LiHMDS, slabě interagující TMEDA snadno disociuje z LiHMDS, což vede ke vzniku dimerů LiHMDS, které jsou reaktivnější. U LiHMDS tedy TMEDA nezvyšuje reaktivitu snížením stupně shlukování.[26] TEDA také na rozdíl od jednoduchých alkyllithných sloučenin nezpůsobuje deagregaci lithio-acetofenolátu rozpuštěného v tetrahydrofuranu (THF).[5][27] Přidání HMPA k amidům lithia, jako jsou LiHMDS a LDA, v THF často vede ke vzniku směsi shluků monomerů a dimerů. Poměr množství dimerů a monomerů se však nezmění přidáním HMPA a pozorovaný nárůst reaktivity tak není výsledkem deagregace. Mechanismus zvyšování reaktivity těmito látkami je stále předmětem výzkumu.[18]
Reaktivita a využití
Vazba C-Li v organolithných sloučeninách je velmi polární; na atomu uhlíku se tak soustřeďuje většina elektronové hustoty a dochází ke vzniku karbaniontu. Organolithné sloučeniny jsou tak silně zásadité a nukleofilní. Nejběžněji se v syntéze využívají jako nukleofilní činidla, silné zásady pro deprotonaci, k zahájení polymerizace, nebo jako výchozí látky pro přípravu ostatních organokovových sloučenin.
Karbolithiační reakce
U organolithných sloučenin díky jejich nukleofilitě mohou probíhat karbolithiační reakce, kdy je vazba uhlík-lithium naadována na dvojnou nebo trojnou vazbu mezi dvěma uhlíkovými atomy za vzniku nových organolithných sloučenin.[28] Tyto reakce jsou vůbec nejběžněji prováděnými reakcemi organolithných sloučenin. Karbolithiace je důležitá při aniontových polymeračních procesech; n-butyllithium se používá jako katalyzátor zahajující polymeraci styrenu, buta-1,3-dienu, isoprenu či jejich směsí.[29][30]
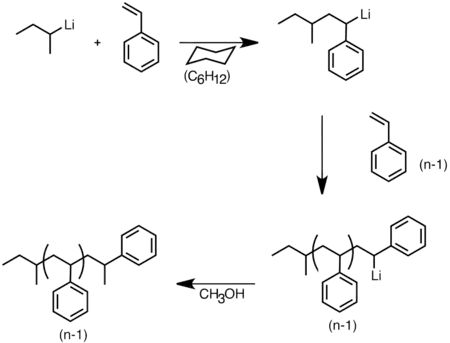 Aniontová polymerace styrenu zahájená sec-butyllithiem
Aniontová polymerace styrenu zahájená sec-butyllithiem
Dalším procesem, při němž se využívá této reaktivity, je tvorba karbocyklických a heterocyklických sloučenin intramolekulární karbolithiací.[28] Tato forma aniontové cyklizace má oproti radikálové cyklizaci několik výhod. Předně je možné, aby vzniklý cyklický organolithný meziprodukt reagoval s elektrofily, což je u radikálových meziproduktů často obtížné. Zadruhé jsou aniontové cyklizace mnohdy, například u 5-hexenyllithných sloučenin, více regio- a stereospecifické než radikálové cyklizace. Intramolekulární karbolithiace umožňuje adici alkyl- a vinyllithných sloučenin na trojné a monoalkylsubstituované dvojné vazby. Tuto adici lzte provést i u aryllithných sloučenin, pokud má vzniknout pětičlenný cyklus. Mezi obtížné varianty intramolekulární karbolithiace patří ty s tvorbou tříčlenných a čtyřčlenných cyklů, jelikož mají příslušné cyklické organolithné meziprodukty často sklon k otevírání cyklů.[28] Lithné sloučeniny vzniklé lithium-halogenovou výměnou se cyklizují na vinyllithné produkty. Tyto látky pak dále reagují s elektrofily za tvorby funkcionalizovaných cyklopentylidenových sloučenin.[31]
Příklad intramolekulární karbolithiační reakce je zobrazen zde:
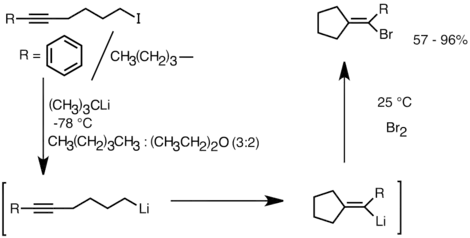 Příklad stereoselektivní intramolekulární karbolithiační reakce
Příklad stereoselektivní intramolekulární karbolithiační reakce
Adice na karbonylové sloučeniny
Nukleofilní organolithné sloučeniny mohou být adovány na elektrofilní dvojné vazby karbonylových skupin za vytvoření vazby uhlík-uhlík. Mohou reagovat s aldehydy či ketony za vzniku alkoholů. Tyto reakce patří převážně mezi polární adice, kdy nukleofilní organolithná sloučenina atakuje karbonyl v ekvatoriálním směru a vzniká axiální alkohol.[32] Selektivitu reakce lze zlepšit přidáním lithných solí jako je například LiClO4.[33]
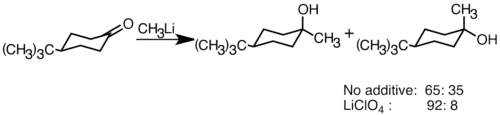 LiClO4 zvyšuje selektivitu reakce t-butyllithia
LiClO4 zvyšuje selektivitu reakce t-butyllithia
U sterickými efekty ovlivněných ketonů použití Grignardových činidel často vede k redukci karbonylové skupiny namísto adice.[32] Alkyllithná činidla jsou vůči této redukci méně náchylná, a mohou být použita na přípravu substituovaných alkoholů.[34] Níže je zobrazen příklad adice ethyllithia na adamanton za vzniku terciárního alkoholu.[35]
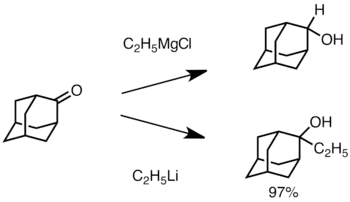 Adice organolithné sloučeniny na adamanton
Adice organolithné sloučeniny na adamanton
Organolithné sloučeniny jsou oproti Grignardovým činidlům lepší také ve schopnosti reagovat s karboxylovými kyselinami na ketony.[32] Tuto reakci lze optimalizovat opatrným kontrolováním množství přidaného organolithného činidla nebo použitím trimethylsilylchloridu k potlačení přebytku organolithného činidla.[36] Běžnějším způsobem syntézy ketonů je adice organolithných sloučenin na Weinrebovy amidy (N-methoxy-N-methyl amidy). Tato reakce poskytuje ketony při přebytku organolithného činidla, a to díky chelataci lithného kationtu mezi N-methoxy- a karbonylový kyslík, čímž vzniká čtyřstěnný meziprodukt, který se v kyselém prostředí dále přeměňuje.[37]
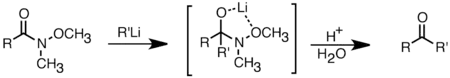 Adice organolithné sloučeniny na Weinrebův amid
Adice organolithné sloučeniny na Weinrebův amid
Organolithné sloučeniny lze také použít k přípravě karboxylových kyselin, a to reakcí s oxidem uhličitým.[38]
Při použití enonových substrátů, kde jsou dvě možná místa, na nichž může proběhnout nukleofilní adice (1,2-adice na karbonylový uhlík nebo 1,4-konjugovaná adice na β uhlík, u většiny reaktivních organolithných sloučenin probíhá přednostně 1,2-adice, je ovšem známo několik způsobů, jak v tomto případě provést konjugovanou adici. Platí, že jelikož je 1,4-adukt termodynamicky upřednostňovaný, tak lze konjugované adice dosáhnout izomerací produktů, obzvláště pokud je lithná sloučenina jen slabým nukleofilem a 1,2-adice je vratná. Další možností je přidání donorových ligandů do reakční směsi, přičemž vznikají heteroatomem stabilizované organolithné sloučeniny, které snáze podléhají 1,4-konjugované adici než 1,2-adici. Za nepřítomnosti donorového ligandu je lithný kation těsně koordinován na atom kyslíku, ovšem je-li rozpuštěn v HMPA, tak se tato koordinace zeslabuje. Tuto metodu většinou nelze použít k ovlivnění regioselektivity alkyl- a aryllithných činidel.[39][40]
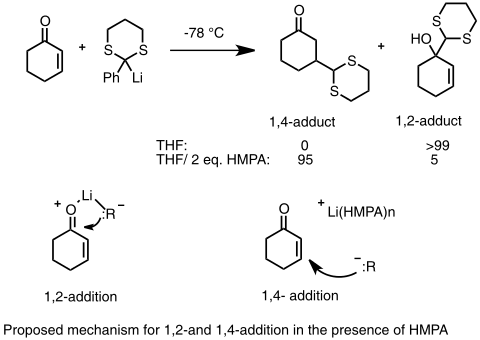 1,4 adice a 1,2 adice
1,4 adice a 1,2 adice
Pomocí organolithných sloučenin lze rovněž, často za přítomnosti chirálních ligandů, provádět enantioselektivní nukleofilní adice na karbonylové sloučeniny a jejich deriváty. Takovéto reakce mají značné využití při průmyslové výrobě léčiv. Příkladem může být syntéza látky Efavirenz, inhibitoru reverzní transkriptázy používaného proti HIV. Acetylid lithný je adován na prochirální keton za vzniku chirálního alkoholu. Struktura aktivního reakčního meziproduktu v roztoku byla určena pomocí NMR spektroskopie a v pevném skupenství pomocí rentgenové krystalografie a bylo zjištěno, že tato látka vytváří krychlový 2:2 tetramer.[41]
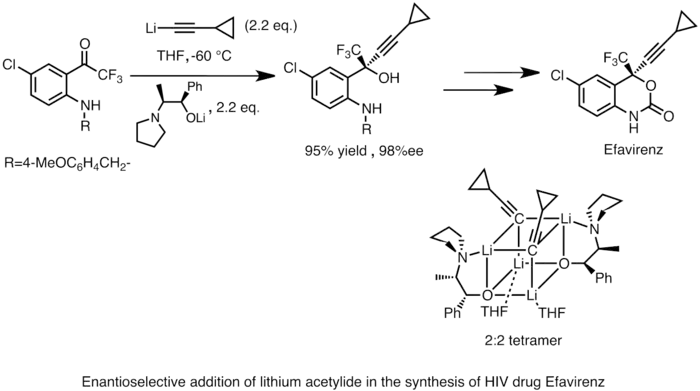
SN2 reakce
Organolithná činidla mohou sloužit jako nukleofily a podstupovat SN2 reakce s alkyl- a allylhalogenidy.[42] I když jsou považovány za reaktivnější alkylační činidla oproti Grignardovým činidlům, tak je jejich využití stále omezené kvůli vedlejším reakcím jako jsou radikálové reakce nebo výměna kovu a halogenu. Organolithná činidla používaná při SN2 reakcích jsou většinou více stabilizována, méně zásadité a méně shluknuté; patří k nim například heteroatomy stabilizovaná aryl- nebo allyllithné sloučeniny.[5] Rychlost a výtěžnost těchto reakcí lze zvýšit přidáním HMPA a reaktivita aryllithných sloučenin může být vylepšena pomocí draselných alkoxidů.[32] Organolithná činidla mohou též nukleofilně reagovat s epoxidy za vzniku alkoholů.
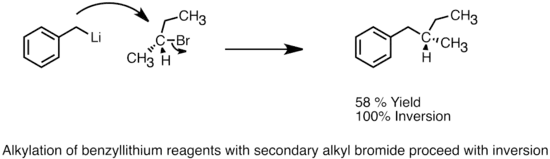 SN2 přeměna provedená pomocí benzyllithia
SN2 přeměna provedená pomocí benzyllithia
Využití organolithných sloučenin jako zásad
Organolithné sloučeniny jsou značně zásadité. Terc-butyllithium s třemi slabě elektronově donorovými alkylovými skupinami, je nejsilnější komerčně dostupnou zásadou (pKa = 53). Tato skutečnost vede k tomu, že kyselostní proton na -OH, -NH a -SH skupinách je často chráněn za přítomnosti organoltithných sloučenin. K běžně používaným lithným zásadám patří alkyllithné sloučeniny jako jsou n-butyllithium a dialkylamidy lithia (LiNR2). Činidla s objemnými substituenty jako jsou například diisopropylamid lithný (LDA) a bis(trimethylsilyl)amid lithný (LiHMDS) jsou pro nukleofilní adici často stericky ovlivněné, a tedy více selektivní k deprotonaci. Lithné dialkylamidy (LiNR2) se často používají na přípravu enolátů a na aldolové reakce.[43] Reaktivita a selektivita těchto zásad je často ovlivněna přítomností dalších iontů a druhem rozpouštědla.
Metalace
Metalace za použití organolithných činidel, také známá jako lithiace nebo lithium-vodíková výměna, se provádí pomocí organolithných, nejčastěji alkyllithných, sloučenin. Dochází k odštěpení protonu a jeho nahrazení lithným kationtem:
- R-H + R'Li → RLi + R'H
K obvyklým lithiačním činidlům patří izomery butyllithia. Terc-butyllithium a sec-butyllithium jsou reaktivnější než n-butyllithium, jsou však také dražší a hůře se skladují.[43]
Lithiace je běžným způsobem přípravy řady organolithných sloučenin. Pozice, na níž k metalaci dojde, je nejvíce ovlivněna kyselostí vazby C-H. Často k&ní dochází na pozici α vzhledem ke skupinám snižujícím elektronovou hustotu, jelikož stabilizují elektronovou hustotu aniontu. Řídicí skupiny u aromatických a heterocyklických sloučenin poskytují místa k regioselektivní metalaci; řízená orthometalace je jednou z důležitých metalačních reakcí. Metalované sulfonové, acylové a α metalované amidové skupiny jsou významnými meziprodukty v chemických syntézách. Metalací allyletherů alkyllithnými sloučeninami nebo LDA vzniká anion na α pozici vzhledem ke kyslíku, a následně může dojít k 2,3-Wittigově přesmyku. Přidáním donorových ligandů jako například TMEDA a HMPA lze zvýšit rychlost metalace a rozšířit spektrum možných substrátů.[44] Chirální organolithné lze získat asymetrickou metalací.[45]
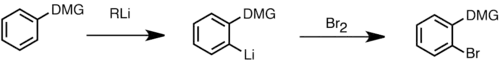 Řízená orthometalace
Řízená orthometalace
Řízená orthometalace je významnou metodou při syntéze regiospecificky substituovaných aromatických sloučenin. Tento způsob lithiace je díky své regioselektivitě často vhodnější než elektrofilní aromatická substituce. Při reakci nejprve organolithné činidlo způsobí deprotonaci na pozici α přímo metalující skupiny na aromatickém kruhu. Touto skupinou je obvykle funkční skupina s heteroatomem, která je Lewisovou zásadou a může se koordinovat na lithný kation, jenž je Lewisovou kyselinou. Tímto vzniká komplexem indukovaný efekt blízkosti, který řídí deprotonaci na pozici α za vzniku aryllithné sloučeniny, která dále může reagovat s elektrofily. K nejúčinnějším přímo metalujícím skupinám patří amidy, karbamáty, sulfony a sulfonamidy. Jsou to skupiny silně snižující elektronovou hustotu, které zvyšují kyselost alfa protonů v benzenovém jádru. Za přítomnosti dvou takovýxh skupin dochází k metalaci v pozici ortho vzhledem k silnější řídicí skupině, byly však pozorovány i smíšené produkty. U mnoha heterocyklů obsahujících kyselostní protony může rovněž proběhnout ortho metalace. U heterocyklických sloučenin s nízkou elektronovou hustotou se většinou používají lithiumamidové zásady jako je LDA, jelikož alkyllithné sloučeniny vyvolávají u těchto heterocyklů spíše adici než deprotonaci. U některých komplexů arenů s přechodnými kovy, jako je například ferrocen, atom kovu přitahuje elektronovou hustotu z arenu, což vede k vyšší kyselosti aromatických protonů a jejich připravenosti na ortho metalaci.[46]
Použití jako superbáze
Adice draselného alkoxidu na alkyllithnou sloučeninu výrazně navýší její zásaditost.[47] Nejběžnější takové superbáze se připravují adicí KOtBu na butyllithium a často se označují zkratkou „LiCKOR“. Jedná se o značně reaktivní a často i stereoselektivní činidla. Při níže zobrazené reakci LiCKOR vytváří metalací a následnou lithium-kovovou výměnou stereospecifické krotylborátové sloučeniny.[48]
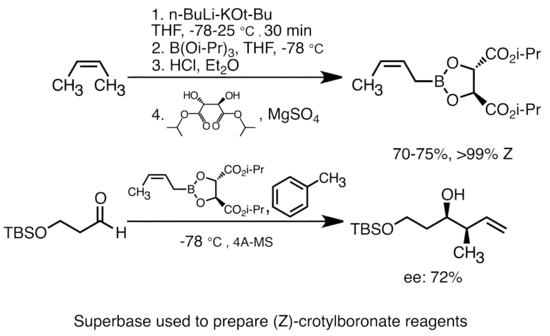 Superbáze
Superbáze
Asymetrické metalace
Enantiomerně obohacené organolithné sloučeniny lze získat asymetrickou metalací prochirálních substrátů. Asymetrická indukce vyžaduje přítomnost chirálního ligandu jako je (−)-spartein.[45] Enantiomerní poměr chirálních organolithných sloučenin je často ovlivněn rozdíly v rychlosti deprotonace. V níže zobrazeném příkladu dochází reakcí N-Boc-N-benzylaminu s n-butyllithiem za přítomnosti (−)-sparteinu ke vzniku jednoho z enantiomerů produktu s vysokým enantiomerním přebytkem. Opačný enantiomer lze získat ransmetalací s trimethylcínchloridem.[49]
-sparteine.png.webp) Asymetrická syntéza s využitím nBuLi a (-)-sparteinu
Asymetrická syntéza s využitím nBuLi a (-)-sparteinu
Tvorba enolátů
Lithné enoláty se dají připravit deptrotonací vazby C-H v α pozici ke karbonylovým skupinám organolithných sloučenin. Používají se jako nukleofily při reakcích vytvářejících vazbu uhlík-uhlík jako jsou například alkylace a aldolová kondenzace. Jsou rovněž významnými meziprodukty při syntéze silylenoletherů.
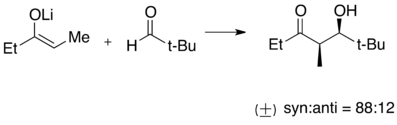 Příklad aldolové reakce s lithným enolátem
Příklad aldolové reakce s lithným enolátem
Tvorbu lithných enolátů lze popsat jako acidobazickou reakci, při níž poměrně kyselý proton z α pozice vzhledem ke karbonylové skupině (pK = 20-28 v DMSO) reaguje s organolithnou zásadou. Používají se silné, nenukleofilní, zásady, obzvláště lithné amidy jako LDA, LiHMDS a LiTMP. Nejčastějšími rozpouštědly při těchto reakcích jsou tetrahydrofuran (THF) a dimethylsulfoxid (DMSO).[50]
Stereochemie a mechanismus tvorby lithných enolátů je mezi chemiky objektem silného zájmu. Stereochemii zde ovlivňuje mnoho faktorů, například sterické efekty, druh rozpouštědla, polární aditiva a druh organolithné zásady. Z mnoha modelů používaných k vysvětlení a předpovídání selektivity ve stereochemii lithných enolátů je Irelandův model.[51]
V tomto modelu reaguje monomerní LDA s karbonylovým substrátem za vzniku cyklického meziproduktu Zimmermanova-Traxlerova typu. Mezi produkty převažuje (E)-enolát, jelikož při vzniku (Z)-enolátového meziproduktu dochází k neupřednostňovaným syn-pentanovým interakcím.[50]
 Irelandův model stereoselektivity lithného enolátu. Zde převažuje (E)-enolát.
Irelandův model stereoselektivity lithného enolátu. Zde převažuje (E)-enolát.
Přidáním polárních aditiv jako například HMPA nebo DMPU dojde k upřednostnění (Z)-enolátu. Podle Irelandova modelu se tyto donorové ligandy koordinují na lithné kationty a interakce lithia s karbonylovým kyslíkem jsou tak omezeny a meziprodukt je slaběji vázán. Podíl (Z)-enolátů se taktéž zvyšuje při použití lithjných zásad s objemnějšími postranními řetězci (jako je například LiHMDS).[50] Mechanismus, kterým tyto látky obracejí stereoselektivitu je však stále neznámý.
Bylo provedeno několik ověření Irelandova modelu, jelikož tento model zobrazuje organolithné sloučeniny v přechodném stavu jako monomery. Ve skutečnosti bylo v roztocích lithných enolátů pozorováno mnoho organolithných shluků a může tak být obtížné určit, který shluk v roztoku je skutečnou reagující látkou.[50]
Lithium-halogenová výměna
Lithium-halogenová výměna je reakce mezi organohalogenidem a organolithnou sloučeninou, obecná rovnice je {R-Li} + R'-X → {R-X} + R'-Li. Objevili ji nezávisle na sobě Henry Gilman a Georg Wittig koncem 30. let 20. století.[52]
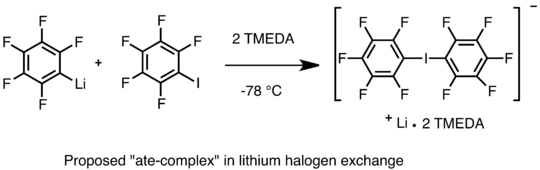
O mechanismu lithium-halogenové výměny se stále spekuluje.[53] Jeden z navržených mechanismů je nukleofilní proces, při němž vzniká vratný meziprodukt v podobě „ate-komplexu“. Byl izolován jodovaný „ate-komplex“ bis(pentafluorofenyl)lithia s TMEDA a podařilo se zjistit i jeho strukturu, a to pomocí rentgenové krystalografie.[54] Tento komplex dále reagoval s elektrofily za vzniku pentafluorofenyljodidu a fenyllithia (C6H5Li).[54] Řada kinetických studií rovněž podporuje nukleofilní mechanismus, kdy karbanion vytvořený z organolithné sloučeniny atakuje halogenový atom arylhalogenidu.[55] Další možný mechanismus spočívá v přesunu jednoho elektronu a tvorbě radikálů; při reakcích sekundárních a terciárních alkyllithných sloučenin s alkylhalogenidy byly EPR spektroskopií detekovány radikály,[56] není však jasné, zda jsou tyto radikály meziprodukty této reakce či nikoliv.[53] Studium mechanismu lithium-halogenové výměny je ztíženo tvorbou shluků organolithných sloučenin.
Lithium-halogenová výměna probíhá velmi rychle. Často je rychlejší než nukleofilní adice a někdy dokonce rychlejší než přenos protonu. V níže uvedeném příkladu je výměna lithia a halogenu v podstatě okamžitá a překonává i přesun protonu z methanolu na terc-butyllithium. Alkenový hlavní produkt se vytváří s více než 90% výtěžností.[57]
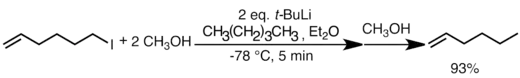
Tato reakce je vhodná pro přípravu nových organolithných sloučenin. Rychlost výměny za jinak stejných podmínek obvykle roste s protonovým číslem halogenu. Alkyl- a arylfluoridy s organolihnými sloučeninami obvykle nereagují. Lithium-halogenová výměna je kineticky ovládána a její rychlost je ovlivňována hlavně stabilitou karbaniontových meziproduktů (sp > sp2 > sp3) organolithných činidel;[32][44] zásaditější terciární organolithné sloučeniny jsou nejreaktivnější a budou reagovat s primárními alkylhalogenidy (obvykle bromidy či jodidy) za vzniku stabilnějších organolithných sloučenin. Lithium-halogenová výměna se tak nejčastěji používá na přípravu vinyl-, aryl- a primárních alkyllithných sloučenin. Lze ji rovněž usnadnit stabilizací karbaniontu přítomností alkoxyskupin nebo heteroatomů; tato metoda je obzvlášť užitečná pro přípravu funkcionalizovaných lithných sloučenin, které by nevydržely tvrdší podmínky nutné při redukci kovovým lithiem.[44] Substráty jako například vinyl halogenidy často procházejí touto výměnou se zachováním stereochemie dvojné vazby.[58]
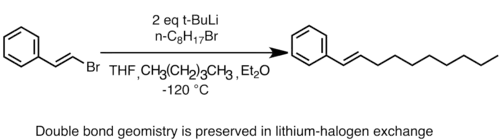 Zachování stereochemie při lithium-halogenové výměně
Zachování stereochemie při lithium-halogenové výměně
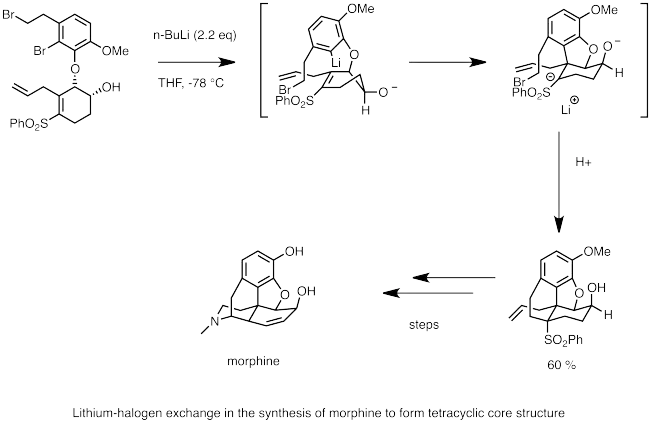
Níže je zobrazen příklad využití lithium-halogenové výměny při syntéze morfinu. N-utyllithium je použito k provedení lithium-halogenové výměny s bromidem. Nukleofilní karbaniontové centrum rychle provádí karbolithiaci dvojné vazby za vzniku aniontu stabilizovaného sousední sulfonovou skupinou. Intramolekulární SN2 reakcí aniontu vzniká cyklický základ molekuly morfinu.[59]
Lithium-halogenová výměna je důležitou součástí Parhamovy cyklizace.[60] V této reakci reaguje arylhalogenid (obvykle jodid či bromid) s organolithnou sloučeninou za vzniku lithiované arenové sloučeniny. Pokud má aren postranní řetězec s elektrofilní částí, pak karbanion připojený na lithium projde intramolekulárním nukleofilním atakem a zcyklizuje se; tato reakce je vhodná například na přípravu heterocyklických sloučenin.[61] V níže zobrazeném příkladě je Parhamova cyklizace použita k přípravě isokyanátu z isoindolinonu, který je následně převeden na nitron. Nitrony dále reagují s radikály a lze je využít jako „spinové pasti“ ke studiu biologických radikálových procesů.[62]
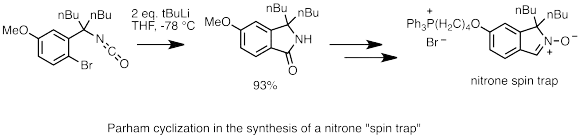 Příklad Parhamovy cyklizace
Příklad Parhamovy cyklizace
Transmetalace
Organolithná činidla se často používají k přípravě dalších, většinou organokovových, sloučenin transmetalací. Organické sloučeniny cínu, mědi, křemíku, boru, fosforu a síry a také organické sloučeniny ceru se často připravují reakcí organolithného činidla s odpovídajícím elektrofilem, například:
- R-M + n-BuLi → RLi + n-BuM
Mezi obvyklé druhy trasmetalace patří výměny typuLi/Sn, Li/Hg a Li/Te, které probíhají rychle i při nízkých teplotách.[43] Výhoda Li/Sn výměny spočívá v tom, že u trialkylstannanových meziproduktů probíhá několik vedlejších reakcí a vzniklé n-Bu3Sn vedlejší produkty nereagují s alkyllithnými sloučeninami.[43] V následujícím případě vinylstannan, připravený z terminálního alkenu, vytváří vinyllithium transmetalací s n-BuLi:[63]
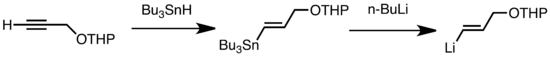 Li/Sn výměna
Li/Sn výměna
Organolithné sloučeniny lze také použít na přípravu organozinkových sloučenin transmetalací se zinečnatými solemi.[64]
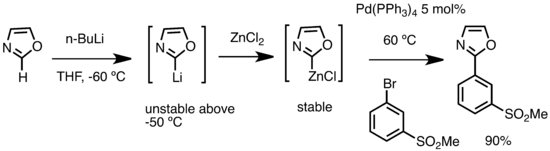 Příprava organozinkové sloučeniny z alkyllithného činidla
Příprava organozinkové sloučeniny z alkyllithného činidla
Reakcí alkyllithných sloučenin s měďnými halogenidy se dají připravit lithné diorganokupráty. Vzniklé sloučeniny jsou obecně méně reaktivní vůči aldehydům a ketonům než organolithná či Grignardova činidla.[65]
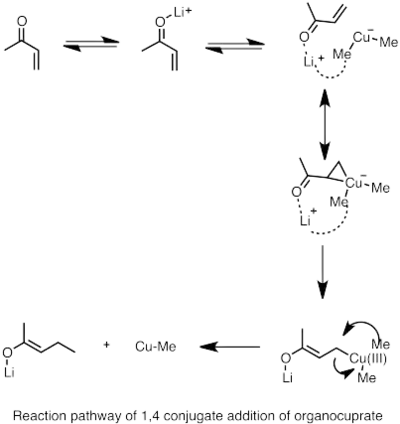 1,4-kuprátová adice
1,4-kuprátová adice
Příprava a výroba
Většina jednodušších alkyllithných činidel a běžnějších lithných amidů je komerčně dostupná v řadě roztoků o různých koncentracích. Lze je rovněž připravit v laboratoři. Níže jsou popsány některé obvyklé způsoby přípravy organolithných sloučenin.
Reakce s kovovým lithiem
Redukcí alkyl- či arylhalogenidů kovovým lithiem lze získat jednoduché alkyl- a aryllithné sloučeniny,[32] obecně:
- R-X + 2 Li → R-Li + Li-X
Průmyslová výroba organolithných sloučenin se provádí za využití výše uvedeného postupu s přídavkem alkylchloridu a kovového lithia obsahujícího 0,5 až 2 % sodíku. Reakce probíhá radikálovým mechanismem, který je iniciován a urychlován sodíkem.[66] Tento proces je značně exotermní. Níže je zobrazen příklad přípravy funkcionalizované organolithné sloučeniny za použití lithia jako redukčního činidla:[67] Někdy se do lithia, ve formě jemného prášku, přidávají některé katalyzátory, jako například naftalen nebo 4,4’-di-t-butylbifenyl (DTBB). Kovovým lithiem lze redukovat i organosulfidy. Redukce organosulfidů je užitečná pro přípravu funkcionalizovaných organolithných činidel, jako jsou alfa-lithio ethery, sulfidy a silany.[68]
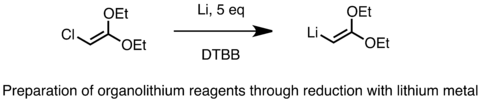 Reduction with Li metal
Reduction with Li metal
Metalace
Dalším způsobem přípravy organolithných sloučenin je metalace (lithium-vodíková výměna). Pozice, na které k ní dojde, je ovlivňována relativní kyselostí vodíkových atomů.
Metalace je nejběžnější metodou přípravy organolithných sloučenin, jelikož koncový vodíkový atom na sp uhlíku je značně kyselý a snadno se deprotonuje.[32] U aromatických sloučenin je pozice lithiace určována též řídícími efekty vytvářenými substituenty.[69] K nejúčinnějším řídícím substituentovým skupinám patří alkoxyskupiny, amidové skupiny, sulfoxid a sulfonyl; metalace často probíhá na ortho pozici vzhledem k těmto substituentům. U heteroaromatických sloučenin obvykle k metalaci dochází na ortho pozici vzhledem k heteroatomu.[32][69]
Lithium-halogenová výměna
Organolithné sloučeniny lze získat rovněž lithium-halogenovou výměnou.
Na přípravu nových organolithných sloučenin halogenovou výměnou se nejčastěji používají terc-butyllithium a n-butyllithium. Tento postup se většinou využívá k přeměně aryl- a alkenyljodidů a bromidů s sp2 uhlíky na odpovídající organolithné sloučeniny. Reakce probíhá velmi rychle a obvykle se provádí při teplotách -60 až -120 °C.[44]
Transmetalace
Čtvrtou metodou přípravy organolithných sloučenin je transmetalace. Tímto způsobem se dá připravit vinyllithium.
Shapirova reakce
V Shapirově reakci reagují dva ekvivalenty silné organolithné zásady s p-tosylhydrazonem za vzniku vinyllithia nebo, po odstranění lithia, alkenového produktu.
Skladování
Organolithné sloučeniny jsou značně reaktivní a vyžadují speciální skladovací postupy. Často jde o žíravé a hořlavé látky, někdy i o samozápalné.[70] Alkyllithná činidla také často podléhají tepelnému rozkladu na odpovídající alkylové sloučeniny a hydrid lithný.[71] Organolithné sloučeniny se obvykle skladují při teplotách pod 10 °C. Koncentrace alkyllithných sloučenin se často určuje titrací.[72]
Organolithná činidla reagují s ethery, které se často používají jako rozpouštědla.[73]
| Rozpouštědlo/Teplota | n-BuLi | s-BuLi | t-BuLi | MeLi | CH2=C(OEt)-Li | CH2=C(SiMe3)-Li |
|---|---|---|---|---|---|---|
| THF/-20 °C | 40 min, 360 min | |||||
| THF/20 °C | >15 h | 17 h | ||||
| THF/35 °C | 10 min | |||||
| THF/TMEDA/-20 °C | 55 h | |||||
| THF/TMEDA/ 0 °C | 340 min | |||||
| THF/TMEDA/20 °C | 40 min | |||||
| Diethylether/-20 °C | 480 min | |||||
| Diethylether/0 °C | 61 min | |||||
| Diethylether/20 °C | 153 h | <30 min | 17 dnů | |||
| Diethylether/35 °C | 31 h | |||||
| Diethylether/TMEDA/ 20 °C | 603 min | |||||
| DME/-70 °C | 120 min | 11 min | ||||
| DME/-20 °C | 110 min | 2 min | <<2 min | |||
| DME/0 °C | 6 min |
Odkazy
Související články
Externí odkazy
 Obrázky, zvuky či videa k tématu Organolithné sloučeniny na Wikimedia Commons
Obrázky, zvuky či videa k tématu Organolithné sloučeniny na Wikimedia Commons
Reference
V tomto článku byl použit překlad textu z článku Organolithium reagent na anglické Wikipedii.
- ZABICKY, Jacob. PATAI'S Chemistry of Functional Groups.. [s.l.]: John Wiley & Sons, Ltd, 2009. ISBN 9780470682531. DOI 10.1002/9780470682531.pat0304. Kapitola Analytical aspects of organolithium compounds. (anglicky)
- EISCH, John J. Henry Gilman: American Pioneer in the Rise of Organometallic Chemistry in Modern Science and Technology†. Organometallics. 2002, s. 5439–5463. ISSN 0276-7333. DOI 10.1021/om0109408. (anglicky)
- The Chemistry of Organolithium Compounds (2 parts).. Redakce Rappoport, Z.. [s.l.]: John Wiley & Sons, Ltd, 2004. ISBN 978-0-470-84339-0. (anglicky)
- STEY, Thomas; STALKE, Dietmar. PATAI'S Chemistry of Functional Groups.. [s.l.]: John Wiley & Sons, Ltd, 2009. ISBN 9780470682531. DOI 10.1002/9780470682531.pat0298. Kapitola Lead structures in lithium organic chemistry. (anglicky)
- REICH, Hans J. Role of Organolithium Aggregates and Mixed Aggregates in Organolithium Mechanisms. Chemical Reviews. 2013, s. 7130–7178. DOI 10.1021/cr400187u. PMID 23941648. (anglicky)
- STROHMANN, C. Structure Formation Principles and Reactivity of Organolithium Compounds.. Chem. Eur. J.. 2009, s. 3320–3334. Dostupné online. DOI 10.1002/chem.200900041. (anglicky)
- JEMMIS, E.D.; GOPAKUMAR, G. PATAI'S Chemistry of Functional Groups.. [s.l.]: John Wiley & Sons, Ltd, 2009. ISBN 9780470682531. DOI 10.1002/9780470682531.pat0297. Kapitola Theoretical studies in organolithium chemistry. (anglicky)
- Streiwieser, A. Perspectives on Computational Organic Chemistry. J. Org. Chem.. 2009, s. 4433–4446. DOI 10.1021/jo900497s. (anglicky)
- Bickelhaupt, F. M. Covalency in Highly Polar Bonds. Structure and Bonding of Methylalkalimetal Oligomers (CH3M)n (M = Li−Rb; n = 1, 4). J. Chem. Theory Comput.. 2006, s. 965–980. DOI 10.1021/ct050333s. (anglicky)
- POWER, P.P; HOPE H. Isolation and crystal structures of the halide-free and halide-rich phenyllithium etherate complexes [(PhLi.Et2O)4] and [(PhLi.Et2O)3.LiBr].. JACS. 1983, s. 5320–5324. Dostupné online. DOI 10.1021/ja00354a022. (anglicky)
- Williard, P. G.; SALVINO, J. M. Synthesis, isolation, and structure of an LDA-THF complex. Journal of Organic Chemistry. 1993, s. 1–3. DOI 10.1021/jo00053a001. (anglicky)
- HILMERSSON, Goran; GRANANDER, Johan. PATAI'S Chemistry of Functional Groups.. [s.l.]: John Wiley & Sons, Ltd, 2009. ISBN 9780470682531. DOI 10.1002/9780470682531.pat0342. Kapitola Structure and dynamics of chiral lithium amides. (anglicky)
- Collum, D.B. Lithium Diisopropylamide: Solution Kinetics and Implications for Organic Synthesis. Angew. Chem. Int. Ed.. 2007, s. 3002–3017. DOI 10.1002/anie.200603038. (anglicky)
- Sekiguchi, Akira. Lithiosilanes and their application to the synthesis of polysilane dendrimers. Coord. Chem. Rev.. 2000, s. 11–45. DOI 10.1016/S0010-8545(00)00315-5. (anglicky)
- Collum, D. B. Solution Structures of Lithium Enolates, Phenolates, Carboxylates, and Alkoxides in the Presence of N,N,N′,N′-Tetramethylethylenediamine: A Prevalence of Cyclic Dimers. J. Org. Chem.. 2008, s. 7743–7747. DOI 10.1021/jo801532d. (anglicky)
- Reich, H. J. Aggregation and reactivity of phenyllithium solutions. J. Am. Chem. Soc.. 1998, s. 7201–7210. DOI 10.1021/ja980684z. (anglicky)
- McGarrity, J. F.; OGLE, C.A. High-field proton NMR study of the aggregation and complexation of n-butyllithium in tetrahydrofuran. J. Am. Chem. Soc.. 1985, s. 1805–1810. DOI 10.1021/ja00293a001. (anglicky)
- Reich, H. J. What's going on with these lithium reagents. J. Org. Chem.. 2012, s. 5471–5491. DOI 10.1021/jo3005155. (anglicky)
- WARDELL, J.L. Comprehensive Organometallic Chemistry, Vol. 1. Redakce Wilinson, G.. 1st. vyd. New York: Pergamon, 1982. ISBN 0080406084. Kapitola Chapter 2. (anglicky)
- Strohmann, C.,; GESSNER, V.H. Crystal Structures of n-BuLi Adducts with (R,R)-TMCDA and the Consequences for the Deprotonation of Benzene. J. Am. Chem. Soc.. 2008, s. 11719–11725. DOI 10.1021/ja8017187. PMID 18686951. (anglicky)
- Collum, D. B. Lithium Diisopropylamide: Solution Kinetics and Implications for Organic Synthesis. Angew. Chem. Int. Ed.. 2007, s. 3002–3017. DOI 10.1002/anie.200603038. (anglicky)
- CHALK, A.J; HOOGEBOOM, T.J. Ring metalation of toluene by butyllithium in the presence of N,N,N′,N′-tetramethylethylenediamine. J. Organomet. Chem. 1968, s. 615–618. Dostupné online. DOI 10.1016/0022-328x(68)80091-9. (anglicky)
- REICH, H.J; GREEN, D.P. Spectroscopic and Reactivity Studies of Lithium Reagent - HMPA Complexes. JACS. 1989, s. 8729–8731. Dostupné online. DOI 10.1021/ja00205a030. (anglicky)
- WILLIARD, P.G; NICHOLS, M.A. Solid-state structures of n-butyllithium-TMEDA, -THF, and -DME complexes. JACS. 1993, s. 1568–1572. Dostupné online. DOI 10.1021/ja00057a050. (anglicky)
- Collum, D.B. Is N,N,N,N-Tetramethylethylenediamine a Good Ligand for Lithium?. Acc. Chem. Res.. 1992, s. 448–454. DOI 10.1021/ar00022a003. (anglicky)
- Bernstein, M.P.; COLLUM, D.B. Solvent- and substrate-dependent rates of imine metalations by lithium diisopropylamide: understanding the mechanisms underlying krel. J. Am. Chem. Soc.. 1993, s. 8008–8010. DOI 10.1021/ja00071a011. (anglicky)
- SEEBACH, D. Structure and Reactivity of Lithium Enolates. From Pinacolone to Selective C-Alkylations of Peptides. Difficulties and Opportunities Afforded by Complex Structures.. Angew. Chem. Int. Ed.. 1988, s. 1624–1654. Dostupné online. DOI 10.1002/anie.198816241. (anglicky)
- FANANAS, Francisco; SANZ, Roberto. PATAI'S Chemistry of Functional Groups.. [s.l.]: John Wiley & Sons, Ltd, 2009. ISBN 9780470682531. DOI 10.1002/9780470682531.pat0341. Kapitola Intramolecular carbolithiation reactions. (anglicky)
- Heinz-Dieter Brandt, Wolfgang Nentwig1, Nicola Rooney, Ronald T. LaFlair, Ute U. Wolf, John Duffy, Judit E. Puskas, Gabor Kaszas, Mark Drewitt, Stephan Glander "Rubber, 5. Solution Rubbers" in Ullmann's Encyclopedia of Industrial Chemistry, 2011, Wiley-VCH, Weinheim. DOI:10.1002/14356007.o23_o02
- BASKARA, D.; MULLER, A.H. Controlled and living polymerizations: From mechanisms to applications. Weinheim, Germany: Wiley-VCH Verlag GmbH & Co. KGaA,, 2010. ISBN 9783527629091. DOI 10.1002/9783527629091.ch1. Kapitola Anionic Vinyl Polymerization. (anglicky)
- Bailey, W.F. Preparation and facile cyclization of 5-alkyn-1-yllithiums. Tetrahedron Lett.. 1989, s. 3901–3904. DOI 10.1016/S0040-4039(00)99279-7. (anglicky)
- CAREY, Francis A. Advanced Organic Chemistry: Reaction and Synthesis Pt. B. Kindle. vyd. [s.l.]: Springer, 2007. ISBN 978-0-387-44899-2. Kapitola Organometallic compounds of Group I and II metals. (anglicky)
- ASHBY, E.C.; NODING, S.R. The effects of added salts on the stereoselectivity and rate of organometallic compound addition to ketones. J. Org. Chem.. 1979, s. 4371–4377. Dostupné online. DOI 10.1021/jo01338a026. (anglicky)
- YAMATAKA, Hiroshi. PATAI'S Chemistry of Functional Groups.. [s.l.]: John Wiley & Sons, Ltd, 2009. ISBN 9780470682531. DOI 10.1002/9780470682531.pat0310. Kapitola Addition of organolithium reagents to double bonds. (anglicky)
- Landa, S. Über adamantan und dessen derivate IX. In 2-stellung substituierte derivate. Czech. Chem. Commun.. 1967, s. 570–575. DOI 10.1135/cccc19670570. (anglicky)
- Rubottom, G.M.; KIM, C. Preparation of methyl ketones by the sequential treatment of carboxylic acids with methyllithium and chlorotrimethylsilane. J. Org. Chem.. 1983, s. 1550–1552. DOI 10.1021/jo00157a038. (anglicky)
- Zadel, G.; BREITMAIER, E. A One-Pot Synthesis of Ketones and Aldehydes from Carbon Dioxide and Organolithium Compounds. Angew. Chem. Int. Ed.. 1992, s. 1035–1036. DOI 10.1002/anie.199210351. (anglicky)
- Ronald, R.C. Methoxymethyl ethers. An activating group for rapid and regioselective metalation. Tetrahedron Lett.. 1975, s. 3973–3974. DOI 10.1016/S0040-4039(00)91212-7. (anglicky)
- Hunt, D.A. Michael addition of organolithium compounds. A Review,. Org. Prep. Proc. Int.. 1989, s. 705–749. DOI 10.1080/00304948909356219. (anglicky)
- Reich, H. J.; SIKORSKI, W. H. Regioselectivity of Addition of Organolithium Reagents to Enones: The Role of HMPA. J. Org. Chem.. 1999, s. 14–15. DOI 10.1021/jo981765g. (anglicky)
- Collum, D.B. NMR Spectroscopic Investigations of Mixed Aggregates Underlying Highly Enantioselective 1,2-Additions of Lithium Cyclopropylacetylide to Quinazolinones. J. Am. Chem. Soc.. 2001, s. 9135–9143. DOI 10.1021/ja0105616. (anglicky)
- Sommmer, L.H.; KORTE, W. D. Stereospecific coupling reactions between organolithium reagents and secondary halides. J. Org. Chem.. 1970, s. 22–25. DOI 10.1021/jo00826a006. (anglicky)
- "Organolithium Reagents Reich, H.J. 2002 http://www.chem.wisc.edu/areas/reich/handouts/lireagents/orgli-primer.pdf
- The Preparation of Organolithium Reagents and Intermediates Leroux.F., Schlosser. M., Zohar. E., Marek. I., Wiley, New York. 2004. ISBN 978-0-470-84339-0
- HOPPE, Dieter; CHRISTOPH, Guido. PATAI'S Chemistry of Functional Groups.. [s.l.]: John Wiley & Sons, Ltd, 2009. ISBN 9780470682531. DOI 10.1002/9780470682531.pat0313. Kapitola Asymmetric deprotonation with alkyllithium– (−)-sparteine. (anglicky)
- CLAYDEN, Jonathan. PATAI'S Chemistry of Functional Groups.. [s.l.]: John Wiley & Sons, Ltd, 2009. ISBN 9780470682531. DOI 10.1002/9780470682531.pat0306. Kapitola Directed metallization of aromatic compounds. (anglicky)
- SCHLOSSER, M. Superbases for organic synthesis. Pure Appl. Chem.. 1988, s. 1627–1634. DOI 10.1351/pac198860111627. (anglicky)
- Roush, W.R. Enantioselective synthesis using diisopropyl tartrate modified (E)- and (Z)-crotylboronates: Reactions with achiral aldehydes. Tetrahedron Lett.. 1988, s. 5579–5582. DOI 10.1016/S0040-4039(00)80816-3. (anglicky)
- Park, Y.S. (−)-Sparteine-Mediated α-Lithiation of N-Boc-N-(p-methoxyphenyl)benzylamine: Enantioselective Syntheses of (S) and (R) Mono- and Disubstituted N-Boc-benzylamines. J. Am. Chem. Soc.. 1996, s. 3757–3758. DOI 10.1021/ja9538804. (anglicky)
- VALNOT, Jean-Yves; MADDALUNO, Jacques. PATAI'S Chemistry of Functional Groups.. [s.l.]: John Wiley & Sons, Ltd, 2009. ISBN 9780470682531. DOI 10.1002/9780470682531.pat0345. Kapitola Aspects of the synthesis, structure and reactivity of lithium enolates. (anglicky)
- Ireland. R. E. The ester enolate Claisen rearrangement. Stereochemical control through stereoselective enolate formation. J. Am. Chem. Soc.. 1976, s. 2868–2877. DOI 10.1021/ja00426a033. (anglicky)
- GILMAN, Henry; LANGHAM, Wright; JACOBY, Arthur L. Metalation as a Side Reaction in the Preparation of Organolithium Compounds. Journal of the American Chemical Society. 1939, s. 106–109. ISSN 0002-7863. DOI 10.1021/ja01870a036. (anglicky)
- Bailey, W. F.; PATRICIA, J. F. The mechanism of the lithium - halogen Interchange reaction : a review of the literature. J. Organomet. Chem.. 1988, s. 1–46. DOI 10.1016/0022-328X(88)83017-1. (anglicky)
- ; CALABRESE, J. C. Novel hypervalent (10-I-2) iodine structures author = Farnham, W. B.. J. Am. Chem. Soc.. 1986, s. 2449–2451. DOI 10.1021/ja00269a055. (anglicky)
- Rogers, H. R.; HOUK, J. Preliminary studies of the mechanism of metal-halogen exchange. The kinetics of reaction of n-butyllithium with substituted bromobenzenes in hexane solution. J. Am. Chem. Soc.. 1982, s. 522–525. DOI 10.1021/ja00366a024. (anglicky)
- Fischer, H. Electron spin resonance of transient alkyl radicals during alkyllithium-alkyl halide reactions. J. Phys. Chem.. 1969, s. 3834–3838. DOI 10.1021/j100845a044. (anglicky)
- BAILEY, W.F. Metal—halogen interchange between t-butyllithium and 1-iodo-5-hexenes provides no evidence for single-electron transfer. Tetrahedron Lett.. 1986, s. 1861–1864. Dostupné online. DOI 10.1016/s0040-4039(00)84395-6. (anglicky)
- SEEBACH, D; NEUMANN H. Stereospecific preparation of terminal vinyllithium derivatives by Br/Li-exchange with t-butyllithium. Tetrahedron Lett.. 1976, s. 4839–4842. Dostupné online. DOI 10.1016/s0040-4039(00)78926-x. (anglicky)
- Toth, J. E.; HAMANN, P.R.; FUCHS, P.L. Studies culminating in the total synthesis of (dl)-morphine. J. Org. Chem.. 1988, s. 4694–4708. DOI 10.1021/jo00255a008. (anglicky)
- Parham, W.P.; BRADSHER, C.K. Aromatic organolithium reagents bearing electrophilic groups. Preparation by halogen-lithium exchange. Acc. Chem. Res.. 1982, s. 300–305. DOI 10.1021/ar00082a001. (anglicky)
- Sotomayor, N.; LETE, E. Aryl and Heteroaryllithium Compounds by Metal - Halogen Exchange. Synthesis of Carbocyclic and Heterocyclic Systems. Curr. Org. Chem.. 2003, s. 275–300. DOI 10.2174/1385272033372987. (anglicky)
- Quin, C. Synthesis of a mitochondria-targeted spin trap using a novel Parham-type cyclization. Tetrahedron. 2009, s. 8154–8160. DOI 10.1016/j.tet.2009.07.081. (anglicky)
- Corey, E.J.; WOLLENBERG, R.H. Useful new organometallic reagents for the synthesis of allylic alcohols by nucleophilic vinylation. J. Org. Chem.. 1975, s. 2265–2266. DOI 10.1021/jo00903a037. (anglicky)
- Reeder, M.R. An Improved Method for the Palladium Cross-Coupling Reaction of Oxazol-2-ylzinc Derivatives with Aryl Bromides. Org. Process Res. Dev.. 2003, s. 696–699. DOI 10.1021/op034059c. (anglicky)
- Nakamura, E. Reaction Pathway of the Conjugate Addition of Lithium Organocuprate Clusters to Acrolein. J. Am. Chem. Soc.. 1997, s. 4900–4910. DOI 10.1021/ja964209h. (anglicky)
- "Organometallics in Organic Synthesis", Schlosser, M., Ed, Wiley: New York, 1994. ISBN 0-471-93637-5
- Si-Fodil, M. Obtention of 2,2-(diethoxy) vinyl lithium and 2-methyl-4-ethoxy butadienyl lithium by arene-catalysed lithiation of the corresponding chloro derivatives. Synthetic applications. Tetrahedron Lett.. 1998, s. 8975–8978. DOI 10.1016/S0040-4039(98)02031-0. (anglicky)
- COHEN, T; BHUPATHY. M. Organoalkali compounds by radical anion induced reductive metalation of phenyl thioethers. Acc. Chem. Res.. 1989, s. 152–161. Dostupné online. DOI 10.1021/ar00160a006. (anglicky)
- SNIECKUS, V. Directed ortho metalation. Tertiary amide and O-carbamate directors in synthetic strategies for polysubstituted aromatics. Chem. Rev.. 1990, s. 879–933. Dostupné online. DOI 10.1021/cr00104a001. (anglicky)
- SCHWINDEMAN, James A.; WOLTERMANN, Chris J.; LETCHFORD, Robert J. Safe handling of organolithium compounds in the laboratory. Chemical Health and Safety. 2002, s. 6–11. ISSN 1074-9098. DOI 10.1016/S1074-9098(02)00295-2. (anglicky)
- GELLERT, H; ZIEGLER, K. Organoalkali compounds. XVI. The thermal stability of lithium alkyls.. Liebigs Ann. Chem.. 1950, s. 179–185. DOI 10.1002/jlac.19505670110. (anglicky)
- KOFRON, W.G.; BACLAWSKI, L.M. A convenient method for estimation of alkyllithium concentrations. J. Org. Chem.. 1976, s. 1879–1880. Dostupné online. DOI 10.1021/jo00872a047. (anglicky)
- Stanetty, P.; KOLLER, H.; MIHOVILOVIC, M. Directed Ortho-Lithiation of Phenylcarbamic Acid 1,l-Dimethylethyl Ester (N-Boc-aniline). Revision and Improvements. J. Org. Chem.. 1992, s. 6833–6837. DOI 10.1021/jo00051a030. (anglicky)