Organické sloučeniny zinku
Organické sloučeniny zinku jsou sloučeniny obsahující vazby mezi atomy uhlíku a zinku, jejich přípravou a vlastnostmi se zabývá organozinková chemie.[1][2][3][4]
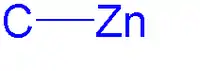
Organozinkové sloučeniny byly jedněmi z prvních připravených organokovových sloučenin. Jsou méně reaktivní než mnohé jiné organokovy, jako například Grignardova a organolithná činidla.
První organozinkovou sloučeninu získal v roce 1848 Edward Frankland, zahříváním jodethyanu s kovovým zinkem vytvořil diethylzinek.[5] Touto reakcí se vytvořila těkavá bezbarvá kapalina samozápalná při styku se vzduchem.
Vzhledem ke své samozápalnosti se organické sloučeniny zinku obvykle připravují za nepřítomnosti vzduchu. Nestálé jsou i za přítomnosti protických rozpouštědel. Mnohdy se připravují těsně před upotřebením a neizolují, řada těchto sloučenin však byla izolována a podrobně prozkoumána.[6]
Organozinkové sloučeniny lze rozdělit do skupin podle počtu uhlíkatých substituentů navázaných na kov.[2][3]
- Diorganozinečnaté sloučeniny (R2Zn): Na zinek jsou navázány dvě alkylové skupiny. Lze je dále dělit podle dalších navázaných skupin.
- Heteroleptické (RZnX): Sloučeniny obsahující elektronegativní nebo monoaniontové ligandy (X), například halogenidy, navázané na zinek a obsahující také alkylové nebo arylové substituenty (R)
- Iontové organozinkové sloučeniny: Dělí se na organozinečnatany (RnZn−) a organozinkové kationty (RZnLn+).
Vazby
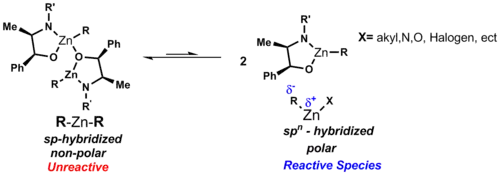
Ve svých komplexech mohou mít zinečnaté ionty několik koordinačních geometrií, nejčastější jsou oktaedrická a tetraedrická. Tuto různorodost lze zdůvodnit elektronovou konfigurací [Ar]3d104s2. Orbital 3d je zcela zaplněn, takže se neprojevují vlivy síly ligandového pole. Koordinační geometrii tak převážně určují elektrostatické a sterické interakce.[2] Organozinkové sloučeniny jsou většinou 2- nebo 3-koordinované, což odpovídá silně donačním vlastnostem karboanionových ligandů.
Obvyklé diorganozinkové komplexy mají vzorec R2Zn. Dialkylzinkové sloučeniny jsou monomerní a alkyly jsou na atom zinku koordinovány lineárně.[7]
Vazby C-Zn jsou polární, polarizované směrem od zinku (elektronegativita 1,65) k uhlíku (elektronegativita 2,5). Dipólový moment je u symetrických lineárních dialkylzinkových sloučenin nulový, což jim dodává rozpustnost v nepolárních rozpouštědlech, jako je cyklohexan. Na rozdíl od jiných binárních alkylů kovů mají diorganozinkové sloučeniny v etherových rozpouštědlech nízkou náchylnost ke tvorbě komplexů. U R2Zn má zinek sp-hybridizované orbitaly.[2]
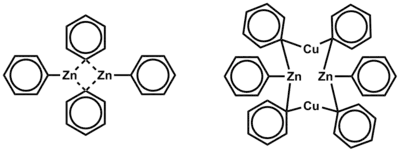
Tyto struktury způsobují, že má zinek dva vazebné d orbitaly a tři nízko položené nevazebné d orbitaly, které mohou vytvořit další vazby. Pokud na zinek nejsou napojeny ligandy dodávající elektrony, tak nelze dosáhnout koordinační nasycenosti, protože má velký atomový poloměr a jen malý nedostatek elektronů. Můstkové alkylové a arylové skupiny se tak v komplexech zinku objevují vzácně, jsou však známy sloučeniny, které je obsahují, jako Ph2Zn. Při navázání halogenu na zinek se zlepšují akceptorové i donorové vlastnosti zinku.[2]
Příprava
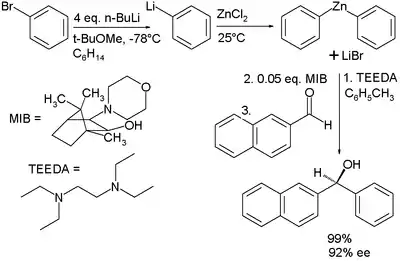
Organozinkové sloučeniny lze vytvořit několika způsoby. Ke komerčně dostupné diorganozinkovým sloučeninám patří dimethylzinek, diethylzinek a difenylzinek. Tyto reaktanty jsou drahé a obtížně se skladují. V jedné studii[8][9] byla aktivní organozinková sloučenina získána z mnohem levnějších organobromidových prekurzorů:
Z kovového zinku
Franklandova syntéza diethylzinku spočívala v reakci jodethanu s kovovým zinkem. Zinek musí být k provedení této reakce aktivován, zde k němu byla přidána měď.[5]
2 EtI + 2 Zn0 → Et2Zn + ZnI2
Dalším druhem aktivovaného zinku je Riekeův zinek, získaný redukcí ZnCl2 draslíkem. Tato forma je vhodná pro reakce jako jsou Negišiovo a Fukujamovo párování. Organozinkové sloučeniny se zde připravují z alkyl- či arylhalogenidů obsahujících skupiny odtahující elektrony, například nitrily a estery.[10][11]


Výměna funkčních skupin
Nejčastějšími reakcemi vedoucími k výměně funkčních skupin navázaných na zinek jsou reakce s halogenidy a borem, katalyzované jodidem měďným (CuI) nebo zásadou. Organoborité reaktanty se zde získávají hydroboracemi a následnými reakcemi jejich produktů s diethylzinkem. Tato příprava poukazuje na vysokou selektivitu většiny reaktivních míst v molekule.[12]
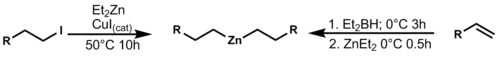
Tento přenos skupin lze využít při allylacích, i při reakcích, jako je Negišiovo párování.[13]
β-silyldiorganozinkové sloučeniny
Jednou z nevýhod alkylací diorganozinkových sloučenin je, že se přesouvá pouze jeden alkyl; řešením může být použití Me3SiCH2-(TMSM),což je nepřenositelná skupina.[14]

Transmetalace
Transmetalace jspou podobné výše uvedeným přeměnám, zinek se zde vyměňuje s jinými kovy, jako jsou například rtuť, lithium a měď. Jako příklad lze uvést reakci difenylrtuti se zinkem za vzniku difenylzinku a rtuti:
HgPh2 + Zn → ZnPh2 + Hg
Výhoda transmetalace tkví v tom, že je možné použít sloučeniny s mnoha různými funkčními skupinami, které snižují reaktivitu a zvyšují selektivitu.[15]
- Při syntéze Maoecrystalu V vzniká řízenou ortho-metalací aryllithná sloučenina, která je následně transmetalována na arylzinkovou. Arylzinková sloučenina je výrazně méně reaktivní než aryllithná a tak lépe strpí přítomnost substituentu u následující reakce s chloroxalacetátem. Estery jsou vůči organozinkovým sloučeninám stabilní.[16]
Organozinkové sloučeniny lze získat přímo z kovového zinku:[17]
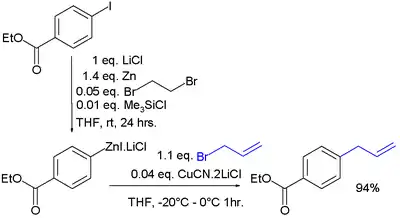
- Zinek se zde aktivuje pomocí 1,2-dibromethanu a trimethylsilylchloridu. Přidává se také chlorid lithný , který s organozinečnatou sloučeninou rychle vytváří rozpustný adukt a odstraňuje ji tak z povrchu kovu.
Reakce
Organické sloučeniny zinku jsou meziprodukty některých organických reakcí.
- Ve Franklandově–Duppaově reakci (objevené roku 1863) reaguje ester kyseliny šťavelové (ROCOCOOR) s halogenalkanem R'X, zinkem a kyselinou chlorovodíkovou za tvorby α-hydroxykarboxylátového esteru, RR'COHCOOR.[19]
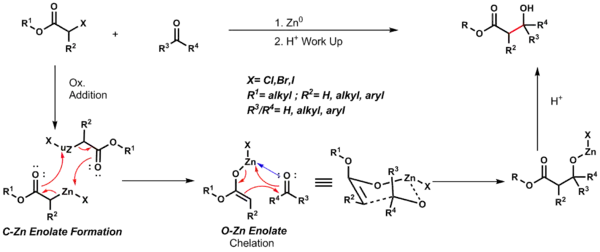
Reformatského reakce
Při Reformatského reakcích se α-halogenestery mění reakcemi s ketony nebo aldehydy na β-hydroxyestery. K protonaci průběžně vytvořeného alkoxidu je potřeba kyselina. Úvodním krokem je oxidační adice zinku na vazbu uhlík-halogen, čímž vznikne uhlíko-zinkový enolát. Tento enolát se následně přesmykne na kyslíko-zinkový. Jakmile se takto vytvoří další karbonyl, tak se další výchozí materiál naváže níže uvedeným způsobem a po protonaci vznikne konečný produkt.[20]
Reformatského reakce mají oproti běžným aldolovým reakcím tyto výhody:
- Umožňují použití i vysoce substituovaných ketonových substrátů.
- Esterenolátový mezprodukt lze získat za přítomnosti enolizovatelných skupin.
- Jsou proveditelné i ve vnitromolekulárně.
Níže je zobrazen šestičlenný cyklický meziprodukt Zimmermanova–Traxleova modelu, kde je skupina R3 menší než R4.[21]
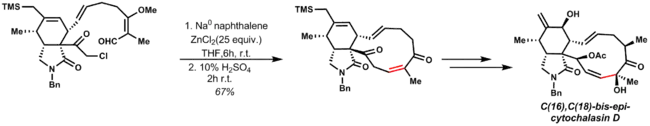
Reformatského reakce byly začleněny do několika totálních syntéz, například u C(16),C(18)-bis-epi-cytochalasinu D:[22]
Do Reformatského reakcí lze také zapojit zinečnaté homoenoláty;[23] jako například při Blaiseových reakcích.[21]
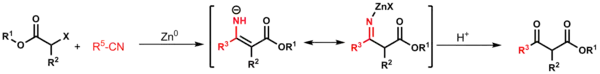
Simmonsovy–Smithovy reakce
Při Simmonsových–Smithových reakcích se připravují cyklopropany z alkenů s využitím dijodmethanu, jako zdroje methylenových skupin, a zinku. Vzniká přitom karbenoidový meziprodukt (jodmethyl)jodid zinečnatý, který reaguje s použitým alkenem za vzniku cyklopropanovaného produktu. Tvorbu aktivní formy zinku lze urychlit pomocí ultrazvuku, protože úvodní reakce probíhá na povrchu kovu.


I když není mechanismus zcela prozkoumán, tak se předpokládá, že organozinečnatý meziprodukt patří mezi karbenoidy. Má být tricentrický a „motýlkovitý“. Jeho tvorbu lze řídit pomocí substituentů, například alkoholových skupin, tak, aby se cyklopropyl navázal na stejnou stranu molekuly. Zinek se obvykle používá ve směsi s mědí.[21]

Methylenace pomocí titanu a zinku
Organozinkové sloučeniny odvozené od dibrommethanu nebo dijodmethanu se mohou elektrofilně adovat na karbonylové sloučeniny za tvorby koncových alkenů.[24]
Mechanismus této reakce je podobný Tebbeově reakci; katalyzovat ji lze mnoha různými Lewisovými kyselinami, jako jsou chlorid titaničitý a trimethylhliník.[25]
Tuto reakci lze použít mimo jiné k zavedení deuteria do molekul za účelem izotopového značkování, případně jako náhradu Wittigovy reakce.
Negišiovo párování
Negišiovým párováním lze vytvořit vazby uhlík-uhlík s využitím organohalogenidu a organozinečnatého halogenidu za přítomnosti katalyzátoru obsahujícího nikl nebo palladium. Organohalogenid může být odvozen od alkenylové, arylové, allylové nebo propargylové skupiny. Byla také popsána párování alkylových sloučenin zinkuas alkylbromidy či alkylchloridy s tvorbou aktivních katalyzátorů z Pd-PEPPSI prekatalyzátorů, které jsou odolné vůči beta-hydridovým eliminacím, jež se u alkylů často vyskytují.[26] Pro transmetalační krok lze použít diorganické i organozinkové halogenidy. I přes nízkou reaktivitu organických sloučenin zinku vůči organickým elektrofilům se jedná o jedny z kovových nukleofilů nejlépe reagujících s palladiem.[27]
Alkylzinkové sloučeniny vyžadují přítomnost alespoň stechiometrického množství halogenidových iontů v roztoku, které umožňují vytváření „zinečnatanů“ typu RZnX
3p=2− před transmetalací na palladiové centrum.[28]
Nejdůležitěší částí katalytického cyklu je transmetalace, kde halogenid zinku vyměňuje organický substituent s atomem halogenu na kovovém centru.
Jako příklad může být uvedena Furstnerova syntéza amfidinolidu T1:[29]
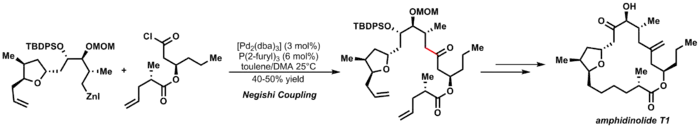
Fukujamovo párování
Fukujamovo párování je reakce aryl-, alkyl, allyl-, nebo α,β-nenasyceného thioesteru katalyzované palladiem. S thioestery zde může reagovat široké apektrum organozinkových sloučenin; produkty jsou ketony. Tento postup se vyznačuje citlivostí na funkční skupiny, jako jsou ketony, acetátové estery, arylhalogenidy a [[[aldehydy]]. Pozorovaná chemoselektivita naznačuje, že tvorba ketonu je jednodušší než oxidační adice na palladium.[30]
Na ukázku zde lze uvést syntézu (+)-biotinu. Zde se do Fukujamova párování zapojuje thiolakton:[31]
-Biotin_Fukuyama_Coupling.png.webp)
Barbierova reakce
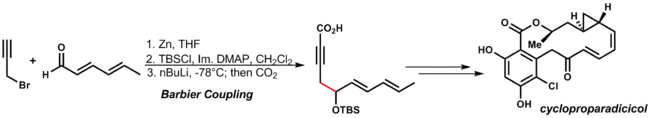
Barbierova reakce spočívá v nukleofilní adici ekvivalentu karboaniontu na karbonyl; podobá se Grignardově reakci. Organozinkové činidlo se vytváří oxidační adicí na alkylhalogenid. Takto lze získat primární, sekiundární i terciární alkoholy pomocí 1,2-adic. Barbierova reakce se provádí v jedné nádobě: organozinková sloučenina se tvoří za přítomnosti karbonylového substrátu. Organozinkové reaktanty použité při této reakci jsou méně citlivé na vodu, takže ji lze provádět ve vodných prostředích. Podobně jako u Grignardovy reakce zde vzniká Schlenková rovnováha, in při níř se tvoří reaktivnější dialkylzinek.[21]
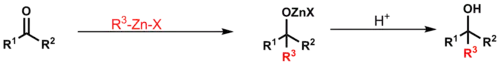
Mechanismus se podobá Grignardově reakci, při níž vzniká alkoxid kovu radikálovým postupným mechanismem přes jednoelektronový přenos nebo soustředěným mechanismem přes cyklický meziprodukt. Jako příklad lze uvést Danishefského syntézu cykloproparadikikolu. Za podmínek, při kterých probíhá adice organozinkové sloučeniny, je možná přítomnost různých skupin na dienonu a alkynu:[32]
Acetylidy zinku
Acetylidy zinku se vytváří přes dialkynylzinkové meziprodukty. Využívají se například na výrobu efedrinu.[33] Lze z nich získat propargylalkoholy. Mohou tak být použity při řadě různých reakcí, jako jsou křížová párování, hydrogenace a pericyklické reakce.[34]
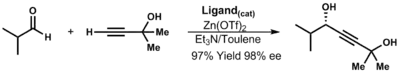
Za nepřítomnosti ligandů probíhá tato reakce velmi pomalu. Pokud jsou přítomny chirální ligandy, tak je reakce velmi rychlá. Rjódži Nojori zjistil, že aktivní je zde komplex s ligandem navázaným na jeden atom zinku.[35]
Diastereoseleltivitu adice organozinkových sloučenin na aldehydy lze předvídat pomocí následujícího modelu, který vyvinuli Rjódži Nojori a David A. Evans:[36]
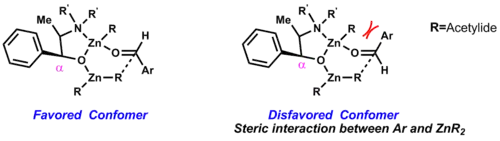
- Stereochemii vzniklého propargylalkoholu je určena α-stereocentrem.
- Sterické efekty mezi aldehydem a ligandem mají menší vliv, ovšem i tak se podílejí na určování převažující konformace.
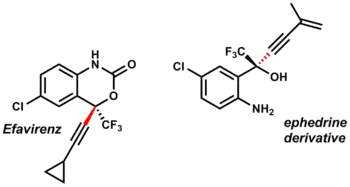
Acetylidy zinku byly použity při přípravách efavirenzu, inhibitoru reverzní transkriptázy u HIV-1 i derivátů efedrinu.[37]
Organozinečnatany
První organozinkový átový komplex (organozinečnatan) popsal James Alfred Wanklyn v roce 1858,[38] připravil jej reakcí sodíku s diethylzinkem:
2 Na + 3 ZnEt2 → 2 NaZnEt3 + Zn
Organozinkové sloučeniny se silnou Lewisovskou kyselostí jsou náchylné k nukleofilním reakcím s alkalickými kovy, jako je sodík, čímž vytvářejí uvedené átové komplexy. Rozlišují se dva druhy organozinečnatanů: tetraorganozinečnatany ([R4Zn]M2), které jsou dianiontové, a monoaniontové triorganozinečnatany ([R3Zn]M). Jejich struktury, určované ligandy, jsou podrobně popsány.[3]
Příprava
Tetraorganozinečnatany, jako [Me4Zn]Li2, mohou být připraveny reakcemi podobnými jako u Me2Zn s MeLi v molárním poměru 1:2. Níže je zobrazena příprava spirocyklických organozinečnatanů:[3]
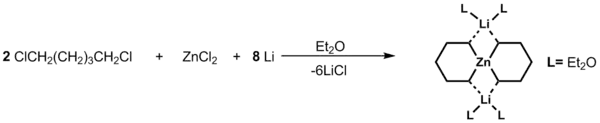
Triorganozinečnatany lze získat reakcemi diorganozinkových sloučenin, například (Me3SiCH2)2Zn, s alkalickými kovy (K), nebo kovy alkalických zemin (Ba, Sr nebo Ca).
Triethylzinečnatany se rozkládají na hydridoethylzinečnatany, protože u nich dochází k betahydridové eliminaci:[39]
2 NaZnEt3 → Na2Zn2H2Et4 + 2 C2H4
Produkt má bitetraedrickou strukturu se společnými vrcholy a obsahuje hydridové můstkové ligandy.
Reakce
Organozinečnatany nejsou zkoumány příliš často, všem vykazují vyšší reaktivitu než neutrální diorganozinky. Využití nacházejí při stereoselektivních alkylacích karbonylových sloučenin a u reakcí otevírajících kruhy. Aryltrimethylzinečnatany se účastní reakcí vytvářejících vazby C-C řízených vanadem.[3]
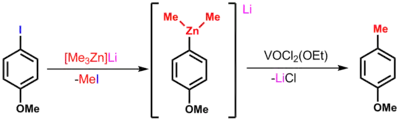
Organozinné sloučeniny
Jsou známy i organické sloučeniny zinku v nižších oxidačních číslech, obsahující vazby Zn–Zn. První takovou sloučeninu, dekamethyldizinkocen, se podařilo připravit v roce 2004.[40]
Odkazy
Reference
V tomto článku byl použit překlad textu z článku Organozinc compound na anglické Wikipedii.
- Paul Knochel; Nicolas Millot; Alain L. Rodriguez; Charles E. Tucker. Organic Reactions. [s.l.]: [s.n.], 2004. ISBN 0471264180. DOI 10.1002/0471264180.or058.02.
- The Chemistry of Organozinc Compounds (Patai Series: The Chemistry of Functional Groups), (Eds. Z. Rappoport and I. Marek), John Wiley & Sons: Chichester, UK, 2006, ISBN 0-470-09337-4.
- Organozinc reagents – A Practical Approach, (Eds. P. Knochel and P. Jones), Oxford Medical Publications, Oxford, 1999, ISBN 0-19-850121-8.
- Synthetic Methods of Organometallic and Inorganic Chemistry Vol 5, Copper, Silver, Gold, Zinc, Cadmium, and Mercury, W.A. Herrmann Ed., ISBN 3-13-103061-5
- E. Frankland, Justus Liebigs Annalen der Chemie, 1849, 71, 171
- Elschenbroich, C. "Organometallics" (2006) Wiley-VCH: Weinheim. ISBN 978-3-527-29390-2
- John Bacsa; Felix Hanke; Sarah Hindley; Rajesh Odedra; George R. Darling; Anthony C. Jones; Alexander Steiner. The Solid State Structures of Dimethylzinc and Diethylzinc. Angewandte Chemie International Edition. 2011, s. 11685–11687. DOI 10.1002/anie.201105099. PMID 21919175.
- Kim Jeung Gon; Patrick J. Walsh. From Aryl Bromides to Enantioenriched Benzylic Alcohols in a Single Flask: Catalytic Asymmetric Arylation of Aldehydes. Angewandte Chemie International Edition. 2006, s. 4175–4178. DOI 10.1002/anie.200600741. PMID 16721894.
- V této přípravě se brombenzen mění reakcí se 4 ekvivalenty n-butyllithia na fenylithium, které je následně transmetalováno reakcí s chloridem zinečnatým za vzniku difenylzinku, jenž dále reaguje s borneolovým (MIB) ligandem a poté s 2-naftylaldehydem za tvorby alkoholu. Společně s difenylzinkem se tvoří chlorid lithný, který by bez přítomnosti MIB způsoboval vznik racemického alkoholu. Tato sůl se odstraní chelatací s tetraethylethylendiaminem (TEEDA) a dosažený enantiomerní přebytek činí 92 %.
- R. D. Rieke. Preparation of Organometallic Compounds from Highly Reactive Metal Powders. Science. 1989, s. 1260–1264. DOI 10.1126/science.246.4935.1260. PMID 17832221. Bibcode 1989Sci...246.1260R.
- Ei-Ichi Negishi. A genealogy of Pd-catalyzed cross-coupling. Journal of Organometallic Chemistry. 2002, s. 34–40. DOI 10.1016/S0022-328X(02)01273-1.
- Falk Langer; Lothar Schwink; Arokiasamy Devasagayaraj; Pierre-Yves Chavant; Paul Knochel. Preparation of Functionalized Dialkylzincs via a Boron−Zinc Exchange. Reactivity and Catalytic Asymmetric Addition to Aldehydes. The Journal of Organic Chemistry. 1996, s. 8229–8243. ISSN 0022-3263. DOI 10.1021/jo961129n. PMID 11667810.
- Naka,H; et al.New J. Chem., 2010, 34, 1700–1706
- Knochel,P.; et al. Angel. Chem. Int. Ed. Engl. 1997, volume 36, 1496-1498
- P. Markies; Gerrit Schat; Otto S. Akkerman; F. Bickelhaupt; Anthony L. Spek. Complexation of diphenylzinc with simple ethers. Crystal structures of the complexes Ph2Zn·glyme and Ph2Zn·diglyme. Journal of Organometallic Chemistry. 1992, s. 1–13. DOI 10.1016/0022-328X(92)80090-K.
- Ping Lu; Zhenhua Gu; Armen Zakarian. Total Synthesis of Maoecrystal V: Early-Stage C–H Functionalization and Lactone Assembly by Radical Cyclization. Journal of the American Chemical Society. 2013, s. 14552–14555. DOI 10.1021/ja408231t. PMID 24047444.
- Arkady Krasovskiy; Vladimir Malakhov; Andrei Gavryushin; Paul Knochel. Efficient Synthesis of Functionalized Organozinc Compounds by the Direct Insertion of Zinc into Organic Iodides and Bromides. Angewandte Chemie International Edition. 2006, s. 6040–6044. DOI 10.1002/anie.200601450. PMID 16900548.
- Aryljodid zinku reguje s allylbromidem v nukleofilní substituci.
- Merck Index. [s.l.]: Merck, 2013. Dostupné online. DOI 10.1021/ja408231t. PMID 24047444. S. 14552–14555.
- Alois Fürstner. Recent Advancements in the Reformatsky Reaction. Synthesis. 1989, s. 57–590. DOI 10.1055/s-1989-27326.
- Kurti, L.; Czako, B. Strategic Applications of Named Reactions in Organic Synthesis; Elsevier: Burlington, 2005.
- E. Vedejs; S. M. Duncan. A Synthesis of C(16),C(18)-Bis-epi-cytochalasin D via Reformatsky Cyclization. The Journal of Organic Chemistry. 2000, s. 6073–6081. DOI 10.1021/jo000533q. PMID 10987942.
- Kumwaijima,I.; et al. J. Am. Chem. 1987, 109, 8056
- Kazuhiko Takai; Yuji Hotta; Koichiro Oshima; Hitosi Nozaki. Wittig-type Reaction of Dimetallated Carbodianion Species as Produced by Zinc Reduction of gem-Polyhalogen Compounds in the Presence of Lewis Acids. Bulletin of the Chemical Society of Japan. 1980, s. 1698–1702. DOI 10.1246/bcsj.53.1698.
- Barry Trost; Ian Fleming; Stuart Schreiber. Comprehensive Organic Synthesis Volume 1: Additions to CX π-Bonds, Part 1. New York: Pergamon Press, 1991. ISBN 9780080405926. DOI 10.1016/B978-0-08-052349-1.00020-2. Kapitola Transformation of the Carbonyl Group into Nonhydroxylic Groups, s. 749–751.
- S. Sase, M. Jaric, A. Metzger, V. Malakhov, P. Knochel, J. Org. Chem., 2008, 73, 7380-7382
- K. C. Nicolaou; Paul G. Bulger; David Sarlah. Palladium-Catalyzed Cross-Coupling Reactions in Total Synthesis. Angewandte Chemie International Edition. 2005, s. 4442–4489. DOI 10.1002/anie.200500368. PMID 15991198.
- L. C. McCann; H. N. Hunter; J. A. C. Cyburne; M. G. Organ. Higher-Order Zincates as Transmetalators in Alkyl-Alkyl Negishi Cross-Coupling. Angewandte Chemie International Edition. 2012, s. 7024–7027. DOI 10.1002/anie.201203547. PMID 22685029.
- Christophe Aïssa; Ricardo Riveiros; Jacques Ragot; Alois Fürstner. Total Syntheses of Amphidinolide T1, T3, T4, and T5. Journal of the American Chemical Society. 2003, s. 15512–15520. DOI 10.1021/ja038216z. PMID 14664598.
- Hidetoshi Tokuyama; Satoshi Yokoshima; Tohru Yamashita; Tohru Fukuyama. A novel ketone synthesis by a palladium-catalyzed reaction of thiol esters and organozinc reagents. Tetrahedron Letters. 1998, s. 3189–3192. DOI 10.1016/S0040-4039(98)00456-0.
- Toshiaki Shimizu; Masahiko Seki. Facile synthesis of (+)-biotin via Fukuyama coupling reaction. Tetrahedron Letters. 2000, s. 5099–5101. DOI 10.1016/S0040-4039(00)00781-4.
- Zhi-Qiang Yang; Xudong Geng; David Solit; Christine A. Pratilas; Neal Rosen; Samuel J. Danishefsky. New Efficient Synthesis of Resorcinylic Macrolides via Ynolides: Establishment of Cycloproparadicicol as Synthetically Feasible Preclinical Anticancer Agent Based on Hsp90 as the Target. Journal of the American Chemical Society. 2004, s. 7881–7889. DOI 10.1021/ja0484348. PMID 15212536.
- Li, Z.; Upadhyay, V.; DeCamp, A. E.; DiMichele, L.; Reider, P. J. Synthesis 1999, 1453-1458.
- Kenso Soai; Seiji Niwa. Enantioselective addition of organozinc reagents to aldehydes. Chemical Reviews. 1992, s. 833–856. DOI 10.1021/cr00013a004.
- Ryoji Noyori; Masato Kitamura. Enantioselective Addition of Organometallic Reagents to Carbonyl Compounds: Chirality Transfer, Multiplication, and Amplification. Angewandte Chemie International Edition in English. 1991, s. 49–69. DOI 10.1002/anie.199100491.
- D. Evans. Stereoselective organic reactions: Catalysts for carbonyl addition processes. Science. 1988, s. 420–426. DOI 10.1126/science.3358127. PMID 3358127. Bibcode 1988Sci...240..420E.
- Thompson, A. S.; Corley, E. G.; Huntington, M. F.; Grabowski, E. J. J. Tetrahedron Lett. 1995, 36, 8937-8940
- J. A. Wanklyn. Ueber einige neue Aethylverbindungen, welche Alkalimetalle enthalten. Liebigs Annalen. 1858, s. 67–79. Dostupné online. DOI doi = 10.1002/jlac.18581080116.
- Anders Lennartson; Mikael Håkansson; Susan Jagner. Facile Synthesis of Well-Defined Sodium Hydridoalkylzincates(II). Angewandte Chemie International Edition. 2007, s. 6678–6680. DOI 10.1002/anie.200701477. PMID 17665387.
- Stephan Schulz. Low-Valent Organometallics-Synthesis, Reactivity, and Potential Applications. Chemistry: A European Journal. 2010, s. 6416–6428. DOI 10.1002/chem.201000580. PMID 20486240.