Organické sloučeniny mědi
Organické sloučeniny mědi jsou organokovové sloučeniny obsahující vazby mezi atomy uhlíku a mědi.[1][2][3] Používají se jako reaktanty v organické chemii.
První připravenou sloučeninou z této skupiny byl výbušný acetylid měďný, Cu2C2 (Cu-C≡C-Cu), v roce 1859 jej připravil Rudolf Christian Böttger reakcí acetylenu s roztokem chloridu měďného:[4]
- C2H2 + 2 CuCl → Cu2C2 + 2 HCl
Struktura
Organické sloučeniny mědi jsou značně různorodé ve struktuře i reaktivitě, jsou však mnohdy omezeny tím, že měď zde obvykle mívá oxidační číslo I, označované také Cu+. Jako d10 komplexy jsou organoměďné sloučeniny podobné obdobným sloučeninám Ni0, ovšem vzhledem k vyššímu oxidačnímu číslu kovu se méně zapojují do vazeb pí. Organické sloučeniny Cu2+ a Cu3+ se vyskytují jako meziprodukty některých reakcí, ovšem málokdy je lze izolovat, někdy ani nejsou pozorovatelné. Z hlediska geometrie mívají měďná centra symetrické struktury. Obvykle zaujímají jednu z těchto tří koordinačních geometrií: lineární 2-koordinovanou, trigonální 3-koordinovanou nebo tetraedrickou 4-koordinovanou. Organoměďné sloučeniny vytváří komplexy s mnoha měkkými ligandy, jako jsou alkylfosfiny (R3P), thioethery (R2S) a kyanidy (CN−).
Jednoduché komplexy s CO, alkeny a Cp
Měďné soli mohou, i když jen slabě, reagovat s oxidem uhelnatým (CO), jako příklad lze uvést komplex CuCl(CO), jenž je polymerní. Na rozdíl od běžných karbonylů kovů v nich nejsou tak silné vazby pí.[5]
Alkeny se rovněž vážou na měďné sloučeniny, i když i zde jde obvykle o slabé vazby. Navazování ethenu na Cu v bílkovinách má takový význam v biologii rostlin, že se ethen řadí mezi rostlinné hormony. Přítomnost ethenu, detekovaná Cu-proteiny, ovlivňuje zrání a řadu dalších procesů.[6]
I když měď nevytváří metalocen, tak lze získat její polosendvičové sloučeniny, například (η-cyklopentadienyltriethylfosfin)měď.[7]
Alkylové a arylové sloučeniny
Alkyl- a arylměďné sloučeniny
Měďné halogenidy reagují s organolithnými sloučeninami za vzniku příslušných organoměďných sloučenin. Tuto oblast rozvinul Henry Gilman, když v roce 1936 popsal methylměď. Fenylměď lze připravit reakcí fenyllithia s bromidem měďným v diethyletheru. Místo organolithných lze použít i Grignardova činidla.
Gilman také zkoumal dialkylměďnany. Tyto látky je možné získat spojením dvou ekvivalentů RLi s měďnými solemi, případně z oligomerních neutrálních organoměďných sloučeni reakcemi s jedním ekvivalentem organolithného činidla.
Sloučeniny typu [CuRn](n−1)− reagují s kyslíkem a s vodou za vzniku oxidu měďného. Také bývají tepelně nestálé, což lze využít při některých reakcích. Vzhledem k těmto vlastnostem se organoměďné sloučeniny často připravují in situ bez toho, aby byly izolovány. Využití mají v organické syntéze jako alkylační činidla, protože mohou být použity se širším rozmezím funkčních skupin než Grignardovy a organolithné reaktanty. Elektronegativita je mnohem vyšší než u sousedního prvku 12. skupiny, zinku, což omezuje nukleofilitu jejích uhlíkatých ligandů.
Měďné soli vytvářejí s koncovými alkyny acetylidy.
Alkylhalogenidy reagují s organoměďnými sloučeninami za obrácení konfigurace, u alkenylhalogenidů se ovšem konfigurace substrátu zachovává.[8]
Takto reagují organoměďné sloučeniny s arylhalogenidy:

Struktura
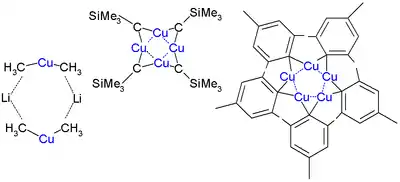
Alkylové a arylové komplexy vytvářejí v krystalové podobě i v roztocích shluky. Tento jev je obzvláště patrný u sloučenin bez elektrického náboje, například těch se stechiometrickým vzorcem typu RCu, jež mají cyklické struktury. Protože musí být na každý atom mědi navázány alespoň dva ligandy, tak organická skupina vytváří můstek; to lze ukázat na struktuře mesitylmědi, jež tvoří pentamer. Cyklickou strukturu má také CuCH2SiMe3, první 1:1 organoměďná sloučenina zkoumaná rentgenovou krystalografií (v roce 1972). Tato sloučenina je poměrně stabilní, protože objemné trimethylsilylové skupiny vytváří tabilizující sterické efekty. Sloučenina vytváří tetramer v podobě osmičlenného kruhu se střídáním vazeb Cu-C. Čtveřice atomů mědi zde zaujímá rovinné uspořádání založené na tricentrických dvouelektronových vazbách. Vzdálenost mezi sousedními atomy Cu je 242 pm, tedy méně než u elementárního kovu (256 pm). U pentamesitylpentamědi se vytváří pětičlenný kruh, podobný (2,4,6-trimethylfenyl)zlatu, a pentafluorfenylměď je tetramerní.[9]
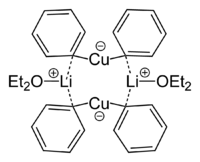
Dimethylměďnan lithný je v diethyletherovém roztoku dimerní s osmičlennou cyklickou strukturou se dvěma atomy lithia spojujícími dvojici methylových skupin. Podobně vytváří dimethylměďnan lithný v pevném skupenství dimerní etherát, [{Li(OEt2)}(CuPh2)]2.[10]
Alkyl- a arylmědité sloučeniny
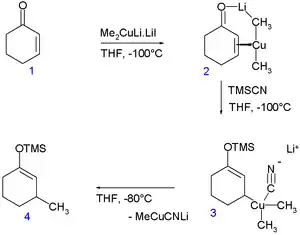
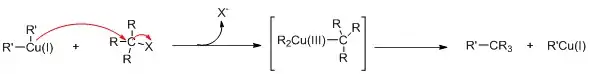
Jinak vzácně se vyskytující měď v oxidačním čísle III se objevuje při konjugované adici Gilmanových činidel na enony.[11]
Experimentem s NMR s rychlým vstřikováním při −100 °C bylo Gilmanovo činidlo Me2CuLi (stabilizované jodidem lithným) zavedeno do cyklohexenonu (1), což umožnilo detekci komplexu Cu— alken 2. Po přidání trimethylsilylkyanidu se vytvořila měditá sloučenina 3 (neomezeně stálá za dané teploty) a po zvýšení teploty na −80 °C se vytvořil produkt konjugované adice 4. Podle in silico experimentů[12] má mít tento meziprodukt čtvercovou rovinnou geometrii s kyanoskupinami v poloze cis oproti methinové skupině cyklohexenylu a antiparalelní vůči methinovému protonu. S jinými ligandy než je kyanoskupina by podle této studie měly vznikat mědité sloučeniny stabilní za pokojové teploty.
Reakce organoměďnanů
Párovací reakce
Při oxidačních párováních se přeměňují acetylidy v Glaserově párování na konjugované alkyny nebo v Castrově–Stephensově párování na arylhalogenidy.
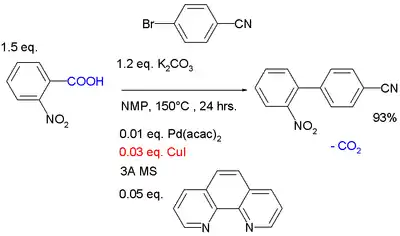
Při redukčních párováních, k jakým patří například Ullmannova reakce, reagují arylhalogenidy se stechiometrickým množstvím mědi, jako příklad lze uvést dekarboxylační párování, kde katalytické množství Cu+ odštěpuje karboxylové skupiny za vzniku arylměďných (ArCu) meziproduktů, současně palladiový katalyzátor přeměňuje arylbromid na organopalladnatý meziprodukt (Ar'PdBr) a pak po transmetalaci z ArPdAr' vznikne biaryl.[13]
Použitými látkami jsou uhličitan draselný (zásada) methylpyrrolidon (rozpouštědlo), acetylacetonát palladnatý a jodid měďný (katalyzátory) a fenantrolin (ligand). Používají se také molekulová síta (na obrázku jako MS)
Neutrální redoxní párování jsou reakce, kdy se spojují koncové alkyny s halogenalkyny za přítomnosti měďné soli; například Cadiotovo–Chodkiewiczovo párování. Lze také provést tepelná párování dvou organoměďných sloučenin.
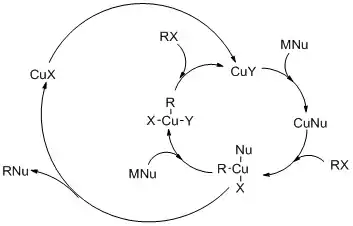
Křížové párovací reakce
Před objevem křížových párování katalyzovaných palladiem byly převážně používány katalyzátory založené na mědi. S palladiem jsou reakce rychlejší a selektivnější. používají se však také Cu katalyzátory, protože jsou levnější a také šetrnější k životnějšímu prostředí.[14]
Reakce R2CuLi s alkylhalogenidy R'-X probíhají takto:
- R2CuLi + R'X → R-R' + CuR + LiX
Součástí mechanismu reakce je oxidační adice alkylhalogenidu na měďnou sloučeninu za vzniku měditého meziproduktu, poté následují redukční eliminace a nukleofilní atak, jenž je krokem určujícím rychlost. Pro substituci jodidem byl navržen mechanismus založený na jednoelektronovém přesunu.

Této reakce se může účastnit mnoho různých elektrofilů ; reaktivita klesá v této řadě:acylchloridy[15] > aldehydy> tosyláty ~ epoxidy > jodidy > bromidy > chloridy > ketony > estery > nitrily >> alkeny
Tento mechanismus je podobný jako u reakcí katalyzovaných palladiem, rozdíl spočívá v tom, že u mědi nastává jednoelektronový přenos.[8]
Karbokuprace
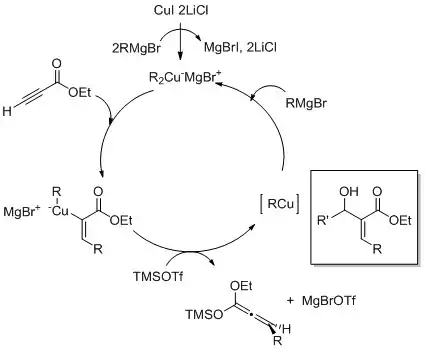
Karbokuprace jsou nukleofilní adiční reakce organoměďných sloučenin (R-Cu) s acetylenem nebo koncovými alkyny za tvorby alkenylměďných produktů (RC=C-Cu);[16]
Jedná se o zvláštní případ karbometalace, nazývaný také Normantova reakce.[17]
Syntetická využití
- Chanovy–Lamovy reakce umožňují vytvářet vazby uhlík-heteroatom v aromatických sloučeninách. Jedná se o párování boronových kyselin a organostannanů nebo siloxanů se substráty obsahujícími skupiny NH- nebo OH-.
- Ulmannovy reakce jsou reakce arylhaliogenidů řízené mědí. Dělí se do dvou druhů:
- Syntézy symetrických biarylových sloučenin
- nukleofilní aromatické substituce
- Sonogaširova reakce, proces, při němž se používá měď i palladium a spojují se aryl- a/nebo vinylhalogenidy s koncovými alkyny.
Tepelné dimerizace probíhají za přítomnosti hydridů mědi, přičemž se zachovává konfigurace organoměďné sloučeniny.[8]
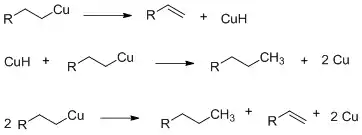
Oxidační dimerizace spočívají v oxidacích dialkylměďnanů na neutrální diakylměďnaté meziprodukty, jež se následně rozkládají na výsledné alkylalkylové dimery. Tato reakce je z hlediska organoněďné sloučeniny i substrátu prvního řádu a dochází při ní k obrácení stereochemie. Mechanismus je pravděpodobně typu SN2.
Redukční činidla
Jako redukční činidla se občas používají hydridy mědi, například Strykerovo činidlo, [(PPh3)CuH]6, které redukuje α,β-nenasycené karbonylové sloučeniny.[19]
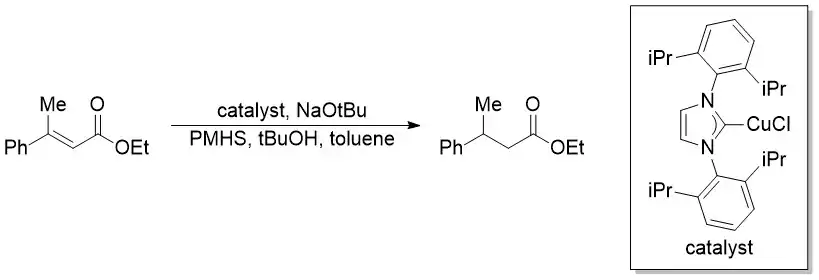
Buchwaldova reakce je asymetrická redukce aktivovaných alkenů katalyzované mědí. Reaktant se připravuje na místě z měďného komplexu NHC. Zdrojem hydridu je zde silan.[20][21]
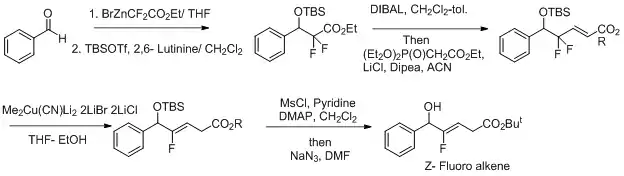
Mědí byla také katalyzována syntéza Z-fluoralkenových izosterů dipeptidů.[22][23] Za účelem zlepšení selektivity této reakce byly použity oxidačně-redukční podmínky.[24] Fluorid funguje jako odstupující skupina a zvyšuje regioselektivitu při přeměně Z-fluoralkenu.
Cu alkylace
Alkylační reakce organoměďných sloučenin jsou obvykle gama-alkylace. Cis-gama atak probíhá, v důsledku sterických efektů, nejlépe u cyklohexylkarbamátu. Reakce je výhodná, pokud se provádí v etherových rozpouštědlech.
Tento postup je velmi účinný u oxidačních párování aminů s alkyl- (například terc-butyl-) a arylhalogenidů.[25]
Vicinální funkcionalizace
Jsou popsány vicinální funkcionalizace spojením karbokupračních reakcí a Mukaijamových aldolových adicí.[26]
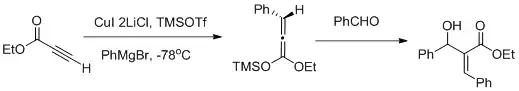
Byla mimo jiné popsána vicinální funkcionalizace α,β-acetylenových esterů karbokuprační reakcí s Mukaijamovou aldolovou adicí, při níž převažuje tvorba Z-aldolu.
Odkazy
Reference
V tomto článku byl použit překlad textu z článku Organocopper compound na anglické Wikipedii.
- Gary H. Posner. An introduction to synthesis using organocopper reagents. [s.l.]: [s.n.], 1980. Dostupné online. ISBN 0-471-69538-6.
- W. A. Herrmann. Synthetic Methods of Organometallic and Inorganic Chemistry. [s.l.]: [s.n.], 1999. ISBN 3-13-103061-5.
- Christoph Elschenbroich. Organometallics. [s.l.]: [s.n.], 2006. ISBN 3-527-29390-6.
- R. C. Böttger. Ueber die Einwirkung des Leuchtgases auf verschiedene Salzsolutionen, insbesondere auf eine ammoniakalische Kupferchlorürlösung. Annalen der Chemie und Pharmacie. 1859, s. 351–362. Dostupné online. DOI 10.1002/jlac.18591090318.
- S. H. Strauss. Copper(I) and Silver(I) Carbonyls. To be or not to be Nonclassical. Journal of the Chemical Society, Dalton Transactions. 2000, s. 1–6. DOI 10.1039/A908459B.
- K. M. Light; J. A. Wisniewski; W. A. Vinyard; M. T. Kieber-Emmons. Perception of the plant hormone ethylene: known-knowns and known-unknowns. Journal of Biological Inorganic Chemistry. 2016, s. 715–728. DOI 10.1007/s00775-016-1378-3. PMID 27456611.
- L. T. J. Delbaere; D. W. McBride; R. B. Ferguson. Crystal structure of π-cyclopentadienyl(triethylphosphine)copper(I), π-C5H5CuP(C2H5)3. Acta Crystallographica B. 1970, s. 515–521. DOI 10.1007/s00775-016-1378-3. PMID 10.1107/S056774087000273X.
- Posner, G. H. 2011. Substitution Reactions Using Organocopper Reagents Organic Reactions 22:2:253–400
- Allan Cairncross; William A. Sheppard; Edward Wonchoba; William J. Guilford; Cynthia B. House; Robert M. Coates. Pentafluorophenylcopper tetramer, a reagent for synthesis of fluorinated aromatic compounds. Organic Syntheses. 1979, s. 122. DOI 10.15227/orgsyn.059.0122.
- N. P. Lorenzen; E. Weiss. Synthesis and Structure of a Dimeric Lithium Diphenylcuprate:[{Li(OEt)2}(CuPh2)]2. Angewandte Chemie International Edition. 1990, s. 300–302. DOI 10.1002/anie.199003001.
- Steven H. Bertz; Stephen Cope; Michael Murphy; Craig A. Ogle; Brad J. Taylor. Rapid Injection NMR in Mechanistic Organocopper Chemistry. Preparation of the Elusive Copper(III) Intermediate1. Journal of the American Chemical Society. 2007, s. 7208–7209. DOI 10.1021/ja067533d.
- Haipeng Hu; James P. Snyder. Organocuprate Conjugate Addition: The Square-Planar "CuIII" Intermediate. Journal of the American Chemical Society. 2007, s. 7210–7211. DOI 10.1021/ja0675346. PMID 17506553.
- L. J. Goossen; G. Deng; L. M. Levy. Synthesis of Biaryls via Catalytic Decarboxylative Coupling. Science. 2006, s. 662–664. DOI 10.1126/science.1128684. PMID 16888137. Bibcode 2006Sci...313..662G.
- I. P. Beletkaya; A. V. Cheprakov. Copper in Cross Coupling Reactions: The Post Ullman Chemistry. Coordination Chemistry Reviews. 2004, s. 2337–2364. DOI 10.1016/j.ccr.2004.09.014.
- Gary H. Posner; Charles E. Whitten. Secondary and Tertiary Alkyl Ketones from Carboxylic Acid Chlorides and Lithium Phenylthio(Alkyl)Cuprate Reagents:tert-Butyl Phenyl Ketone. Organic Syntheses. 2003, s. 122. ISBN 0471264229. DOI 10.1002/0471264180.os055.28.
- Addition of an Ethylcopper Complex to 1-Octyne: (E)-5-Ethyl-1,4-Undecadiene. Organic Syntheses. 1986, s. 1. DOI 10.15227/orgsyn.064.0001. (anglicky)
- J. Normant; M. Bourgain. Synthese stereospecifique and reactivite d' organocuivreux vinyliques. Tetrahedron Letters. 1971, s. 2583. DOI 10.1016/S0040-4039(01)96925-4.
- HENDRIX, AMANDA JOY MUELLER. NOVEL METHODOLOGIES VIA THE CATALYTIC CARBOCUPRATION OF ALKYNOATES AND THE TOTAL SYNTHESIS OF (+)-ASPERGILLIDE B. [s.l.]: [s.n.] Dostupné online. (anglicky)
- John F. Daeuble; Jeffrey M. Stryker. Hexa-μ-hydrohexakis(triphenylphosphine)hexacopper. Encyclopedia of Reagents for Organic Synthesis. 2001. ISBN 0471936235. DOI 10.1002/047084289X.rh011m.
- N. Cox; H. Dang; A. M. Whittaker; G. Lalic. NHC- copper hydrides as chemoselective reducing agents: catalytic reduction of alkynes, alkyl triflates and alkyl halides. Tetrahedron. 2014. DOI 10.1016/j.tet.2014.04.004.
- V. Jurkauskas; J. P. Sadighi; S. L. Buchwald. Conjugate addition of a,b- unsaturated compounds catalyzad by a copper carbene complex. Organic Letters. 2003, s. 2417–2420. DOI 10.1021/ol034560p. PMID 12841744.
- Otaka, A.; Watanabe, H.; Mitsoyama, E.; Yukimasa, A.; Tamamura, H.; Fujii, N. Synthesis of (Z)-fluoroalkene isosteres utilizing organocopper- mediated reduction of gama, gamma- α,β - enoates. Tetrahedron Letters 2001, 42, 285-287.
- Okada, M.; Nakamura, Y. Sago, A.; Hirokawa, H.; Taguchi, T. Stereoselective construction of functionalized (Z)- fluoroalkenes directed to o- depsipeptide isosteres. Tetrahedron Letters 2003, 43, 5845-5847.
- Otaka, A.; Watanabe, H.; Yukimasa, A.; Oishi, S.; Tamamura, H.; Fuji, N. New access to α- substituted (Z)-fluoroalkene dipeptide isosteres utilizing organocopper reagents under redoctive-oxidative alkylation (R-OA) conditions. Tetrahedron Letters 2001, 42, 5443-5446
- H. Yamamoto; K. Marouka. Novel N-alkylation of amines with organocopper reagents. The Journal of Organic Chemistry. 1980, s. 2739–2740. DOI 10.1021/jo01301a048.
- Muller, A. J.; Jennings, M. P. Vicinal Functionalization of propionilate Esters via Tandem Catalytic Carbocupration-Mukaiyama Aldol Reaction sequence Organic Letters 2008, 10, 1649-1652
Literatura
- YAO, B.; LIU, Y.; ZHAO, L.; WANG, D.; WANG, M. Designing a Cu(II)−ArCu(II)−ArCu(III)−Cu(I) Catalytic Cycle: Cu(II)-Catalyzed Oxidative Arene C−H Bond Azidation with Air as an Oxidant under Ambient Conditions. The Journal of Organic Chemistry. 2014, s. 11139–11145. DOI 10.1021/jo502115a. PMID 25350606. (anglicky)
- YAMAMOTO, Y.; YAMAMMOTO, S.; YATAGAI, H.; MARUYAMA, K. Lewis acid mediated reactions of organocopper reagent. A remarkably enhanced regioselective gamma- attack of allylic halides and direct alkylation of allylic alcohols via RCu.BF3. Journal of the American Chemical Society. 1980, s. 2318–2325. DOI 10.1021/ja00527a032. (anglicky)