Totální syntéza chininu
Totální syntéza chininu, léčiva na malárii, byla vyvíjena 150 let. Její vyřešení je považováno za průlom v organické syntéze, i když nebyla nikdy průmyslově využita, protože se chinin získává z přírodních zdrojů. Gilbert Stork oznámil v roce 2001 první stereoselektivní totální syntézu této látky, přestože něco podobného oznámili již Robert Burns Woodward a William von Eggers Doering v roce 1944, kterým se ovšem nepovedlo provést navrženou přeměnu posledního syntetického meziproduktu, chinotoxinu, na chinin. Tato přeměna byla úspěšně provedena až v roce 2007.
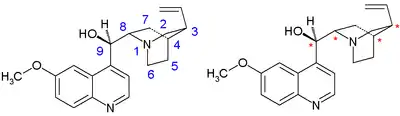
Struktura
Molekula chininu obsahuje aromatické chinolinové jádro, na které je navázána methoxy skupina. Aminová složka má chinuklidinový řetězec a na methylenový můstek je navázán hydroxyl. V poloze 3 se nachází vinylová skupina. Molekula je opticky aktivní, jelikož obsahuje pět stereocenter, kde N1 a C4 tvoří jednu asymetrickou jednotku, což vede k 16 možným stereoizomerům a syntézu ztěžuje.
Historie
- 1817: Pierre Joseph Pelletier a Joseph Caventou provedli první izolaci chininu z chinovníku.
- 1853: Louis Pasteur získal chinotoxin (také nazývaný chinicin) kysele katalyzovanou izomerizací chininu.[1]
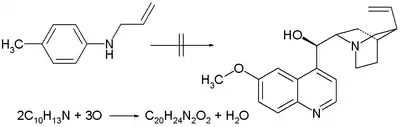
- 1856: William Henry Perkin se pokusil připravit chinin oxidací N-allyltoluidinu, když se domníval, že dva ekvivalenty této látky se souhrnným vzorcem C10H13N se třemi ekvivalenty kyslíku vytvoří jeden ekvivalent C20H24N2O2 (souhrnný vzorec chininu) a a jeden ekvivalent vody.[2] Jeho oxidační reakce dalších toluidinů vedly k objevu mauveinu. Průmyslový význam mauveinu vedl ke vzniku chemického průmyslu.
- 1907: Paul Rabe našel správný strukturní vzorec chininu.[3]
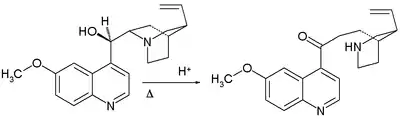
- 1918: Paul Rabe a Karl Kindler vytvořili chinin z chinotoxinu[4] obrácením Pasteurovy reakce. Nedostatek experimentálních důkazů byl téměř o století později předmětem sporů.
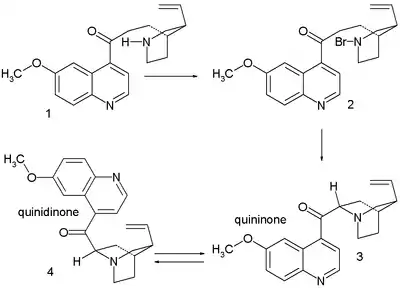
- Prvním krokem je zde reakce bromnanu sodného s chinotoxinem za vzniku N-bromovaného meziproduktu s pravděpodobnou strukturou 2. Následuje oxidace ethoxidem sodným v ethanolu. V zásaditém reakčním prostředí dochází k přeměně chininonu na chinidinon přes enolový meziprodukt a k mutarotaci. Ve třetím kroku se redukuje keton práškovým hliníkem a ethoxidem sodným v ethanolu a vzniká chinin.
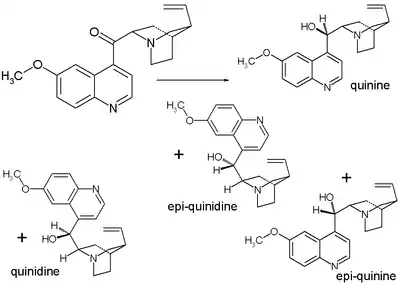
- 1939: Rabe a Kindler prozkoumali vzorek, který zbyl z experimentů z roku 1918 a izolovali a identifikovali chinin a jeho diastereomery chinidin, epichinin a epichinidin.[5]
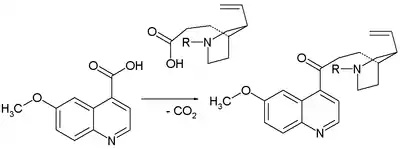
- 1943: Vladimir Prelog a M. Proštenik přeměnili allylpiperidinový derivát homomerochinen na chinotoxin.[6] Homomerochinen byl vytvořen v několika krocích z cinchoninu (podobného chinidinu ale neobsahujícího methoxy skupinu):

- Důležitou součástí tohoto postupu je Claisenova kondenzace:
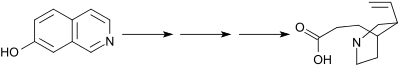
- 1944: Robert Burns Woodward a W. E. Doering popsali syntézu chininu[7] z 7-hydroxyisochinolinu. Přestože tuto práci popsali jako totální syntézu chininu, tak ve skutečnosti vytvořili racemický homomerochinen, poté následovala o několik roků dříve Prelogem popsaná přeměna na chinotoxin (enantiomerně čistý po chirálním rozlišení).
- 1945: Woodward a Doering vydali další článek ohledně chininu.[8]
- 1974: Kijosi Kondo a Fumio Mori připravili racemický vinyl-gama-lakton, který se stal výchozím materiálem při Storkově totální syntéze z roku 2001.[9]
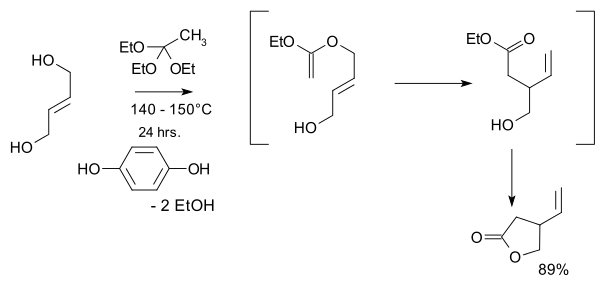
- Výchozími látkami byly trans-but-2-en-1,4-diol a ethylorthoacetát; hlavnímn krokem byl Claisenův přesmyk.
- 1988: Byl připraven enantiomerně čistý lakton pomocí chirálního rozlišení:[10]
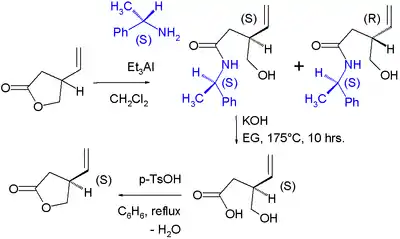
V tomto procesu reagoval racemický lakton v aminolýze s (S)-methylbenzylaminem za přítomnosti triethylhliníku na diastereomerní pár amidů, které byly odděleny sloupcovou chromatografií. S-enantiomer byl převeden zpět na S-lakton hydrolýzou roztokem hydroxidu draselného v ethylenglykolu, po čemž následovalo azeotropní uzavření kruhu.
- 2001: Gilbert Stork popsal stereoselektivní syntézu chininu.[11]
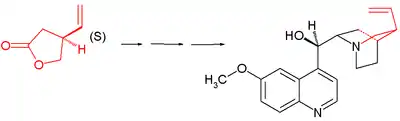
- 2007: Jeffrey I. Seeman v 30stránkovém článku potvrdil správnost Woodwardovy–Doeringovy–Rabeovy–Kindlerovy totální syntézy.[12]
- 2008: Byla povrzena Rabeova příprava chininu z d-chinotoxinu.[13]
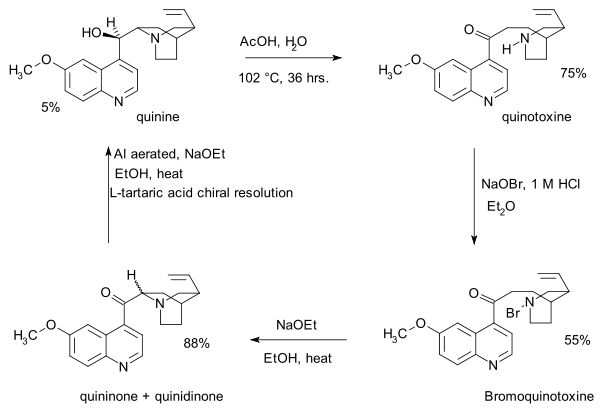
- 2018: Nuno Maulide se svými spolupracovníky popsal totální syntézu chininu s využitím aktivace vazby uhlík-vodík a vytvořil analogy s lepší protimalarickou aktivitou.[14]
Storkova totální syntéza
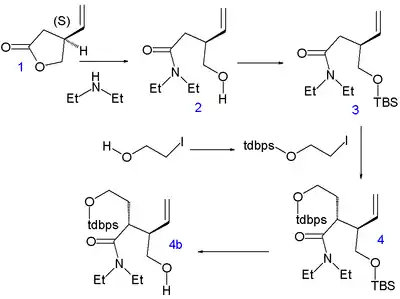
Storkova syntéza chininu začíná u (S)-4-vinylbutyrolaktonu 1, získaného chirálním rozlišením; k tvorbě všech stereogenních center se používá asymetrická indukce a součástí syntézy nejsou žádné asymetrické kroky.
 |
 | |
| Storkova syntéza chininu | Zavedení C8 a dusíku | |
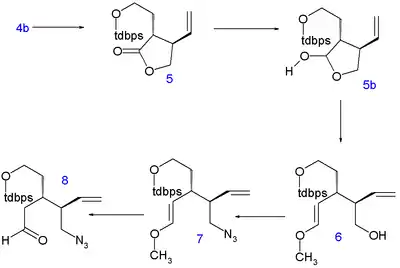
U laktonu proběhlo otevření kruhu diethylaminem za vzniku amidu 2 a jeho hydroxylová skupina byla ochráněna terc-butyldimethylsilyletherem (TBS) (3). Uhlíkové atomy C5 a C6 byly přidány reakcí jodethanolu chráněného terc-butyldifenylsilylovou skupinou (TBDPS) nukleofilní substituční reakcí s diisopropylamidem lithným při −78 °C za vzniku 4 se správnou stereochemií. Oddělením silylové chránicí skupiny kyselinou p-toluensulfonovou se vytvořil alkohol 4b a uzavřením kruhu při azeotropní destilací se obnovila laktonová struktura 5 (přímá alkylace 1 nebyla úspěšná).
Lakton se poté zredukoval diisobutylaluminiumhydridem na laktol 5b a volný aldehyd vstoupil do Wittigovy reakce s methoxymethylentrifenylfosfinem za vzniku enoletheru 6. Hydroxylová skupina byla prostřednictvím Micunobovou reakcí s difenylfosforylazidem nahrazena azidovou 7 a kyselou hydrolýzou se vytvořil azidoaldehyd 8.
 |
 | |
| První uzavírání kruhu | Druhé uzavírání kruhu | |
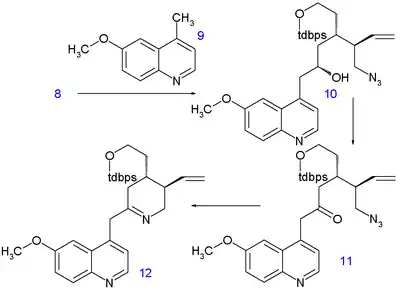
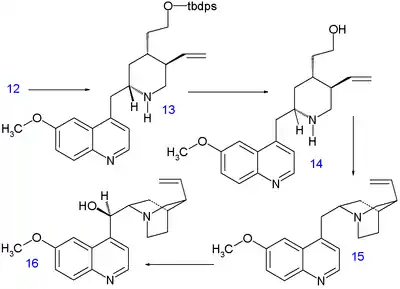
Methylová skupina 6-methoxy-4-methylchinolinu 9 je dostatečně kyselá na to, aby její anion mohl vstoupit do nukleofilní adice za přítomnosti diisopropylamidu lithného na aldehyd 8 za vzniku sloučeniny 10 jako směsi epimerů. Stereochemie zde není důležitá, protože v dalším kroku proběhla Swernova oxidace alkoholu na keton 11. Staudingerovou reakcí s trifenylfosfinem se uzavřel kruh mezi ketonem a azidem a vznikl derivát tetrahydropyridinu 12. Iminový substituent této sloučeniny byl zredukován borohydridem sodným na amin 13 se správnou stereospecifitou. Poté byla odstraněna silylová chránicí skupina kyselinou fluorovodíkovou za tvorby alkoholu 14 a následovala aktivace navázáním mesylové odstupující skupiny reakcí s methansulfonylchloridem v pyridinem, což umožnilo uzavření třetího kruhu 15. V posledním kroku byla na C9 napojena hydroxylová skupina oxidací hydridem sodným v dimethylsulfoxidu a kyslíkem; poměr chininu a epichininu v produktech byl 14:1.
Woodwardova–Doeringova syntéza
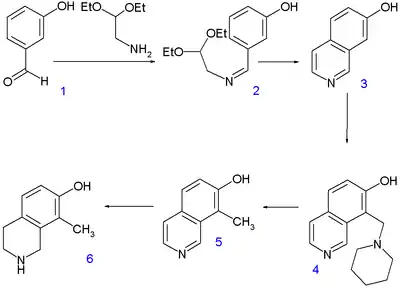
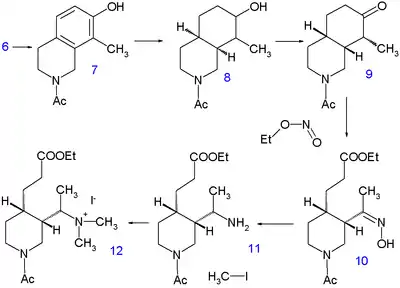
Woodwardova–Doeringova syntéza zahrnuje vytvoření chinuklidinového řetězce z 7-hydroxyisochinolinu (známého od roku 1895) 3 přeměnou stabilního aromatického heterocyklu na plně nasycený bicyklický kruh ve dvou krocích.
 |
 | |
| Woodwardova-Doeringova syntéza chininu I | Woodwardova-Doeringova syntéza chininu II | |
Prvním krokem je kondenzace 3-hydroxybenzaldehydu 1 s diacetalem aminoacetaldehydu za vzniku iminu 2, po níž následuje cyklizace v koncentrované kyselině sírové. Isochinolin 3 byl následně alkylován další kondenzací s formaldehydem a piperidinem a produkt izolován jako sodná sůl 4.
 |
|
| Woodwardova-Doeringova syntéza chininu III | |
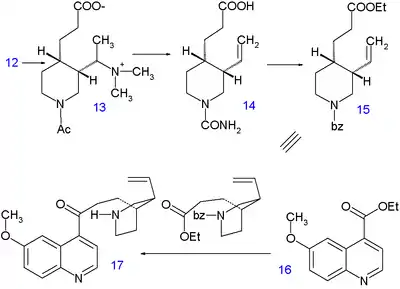
Hydrogenace piperidinu methoxidem sodným v methanolu po dobu 10 hodin při 220 °C uvolnila methylovou skupinu u 5. Druhou hydrogenaci, katalyzovanou Adamsovým katalyzátorem v kyselině octové vznikl tetrahydroisochinolin 6. Další hydrogenace neproběhla, dokud nebyla aminová skupina acylována acetanhydridem v methanolu, sloučenina 7 ovšem prošla opětovnou hydrogenací Raneyovým niklem v ethanolu za 150 °C a vysokého tlaku na dekahydroisochinolin 8. Směs cis- a trans izomerů se následně zoxidovala kyselinou chromovou v kyselině octové na keton 9. Pouze cis-izomer vykrystalizoval a byl použit v dalším kroku, otevírání kruhu ethylnitritem a ethoxidem sodným v ethanolu za vzniku meziproduktu 10 obsahujícího esterovou a oximovou skupinu. Oxim byl hydrogenován na amin 11 platinou v kyselině octové a takto vytvořená sloučenina prošla alkylací jodmethanem za tvorby kvartérní amonné soli 12 a následně the betainu 13 po reakci s oxidem stříbrným.
Vinylová skupina se vytvořila Hofmannovou eliminací za použití vodného roztoku hydroxidu sodného při 140 °C. Došlo přitom k hydrolýze esteru i amidu, nebyl však izolován amin, nýbrž močovina 14, a to po reakci s kyanatanem draselným. V následujícím kroku byla karboxylová kyselina esterifikována ethanolem a močovinová skupina nahrazena benzoylovou. Konečným krokem byla Claisenova kondenzace 15 s ethylchininátem 16, kde po přidání kyseliny vznikl racemický chinotoxin 17. Požadovaný enantiomer byl získán chirálním rozlišením pomocí chirálního dibenzoylesteru kyseliny vinné. Přeměna chinotoxinu na chinin proběhla Rabeovou–Kindlerovou metodou.
Odkazy
Externí odkazy
- Totální syntézy chininu na SynArchive.com
- Historie výzkumu chininu na Harvard.edu
Reference
V tomto článku byl použit překlad textu z článku Quinine total synthesis na anglické Wikipedii.
- Pasteur, L. Comptes rendus de l'Académie des Sciences 1853, 37, 110
- Perkin, W. H. Journal of the Chemical Society 1896, 69, 596
- Rabe, P.; Ackerman, E.; Schneider, W. Berichte der Deutschen Chemischen Gesellschaft 1907, 40, 3655
- Rabe, P.; Kindler, K. Berichte der Deutschen Chemischen Gesellschaft 1918, 51, 466
- P. Rabe, K. Kindler, Berichte der Deutschen Chemischen Gesellschaft B 1939, 72, 263–264.
- Proštenik, M.; Prelog, V. Helvetica Chimica Acta 1943, 26, 1965.
- The Total Synthesis of Quinine R. B. Woodward and W. E. Doering Journal of the American Chemical Society; 1944; 66(5) pp 849 - 849; DOI:10.1021/ja01233a516
- The Total Synthesis of Quinine R. B. Woodward and W. E. Doering Journal of the American Chemical Society; 1945; 67(5) pp 860 - 874; DOI:10.1021/ja01221a051
- SYNTHESIS OF γ-LACTONES BY THE CONDENSATION OF 2-ALKENE-1,4-DIOLS WITH ORTHOCARBOXYLIC ESTERS Kiyosi Kondo and Fumio Mori Chemistry Letters Vol.3 (1974), No.7 pp.741-742 DOI:10.1246/cl.1974.741
- Synthesis and Absolute Configuration of the Acetalic Lignan (+)-Phrymarolin Fumito Ishibashi and Eiji Taniguchi Bulletin of the Chemical Society of Japan Vol.61 (1988), No.12 pp.4361-4366 DOI:10.1246/bcsj.61.4361
- The First Stereoselective Total Synthesis of Quinine Gilbert Stork, Deqiang Niu, A. Fujimoto, Emil R. Koft, James M. Balkovec, James R. Tata, and Gregory R. Dake Journal of the American Chemical Society; 2001; 123(14) pp 3239 - 3242; DOI:10.1021/ja004325r.
- Review: The Woodward-Doering/Rabe-Kindler Total Synthesis of Quinine: Setting the Record Straight Jeffrey I. Seeman Angewandte Chemie International Edition 2007, 46, 1378–1413 DOI:10.1002/anie.200601551 PMID 17294412
- Communication Rabe Rest in Peace: Confirmation of the Rabe–Kindler Conversion of d-Quinotoxine to Quinine: Experimental Affirmation of the Woodward–Doering Formal Total Synthesis of Quinine Aaron C. Smith, Robert M. Williams Angewandte Chemie International Edition 2008, 47, 1736–1740 DOI:10.1002/anie.200705421
- C–H Activation Enables a Concise Total Synthesis of Quinine and Analogues with Enhanced Antimalarial Activity D. H. O'Donovan et al. Angewandte Chemie International Edition 2018 DOI:10.1002/anie.201804551