Stereoselektivita
Stereoselektivita[1] je vlastnost některých chemických reakcí, při kterých se z reaktantu tvoří nerovnoměrná směs stereoizomerů během stereospecifické tvorby nového stereocentra nebo nestereospecifické přeměny již existujícího.[2] Selektivita vzniká v důsledku sterických a/nebo elektronových efektů v průběhu reakčního mechanismu. Míra stereoselektivity se může lišit, nikdy však není úplná, protože rozdíl v aktivačních energiích obou mechanismů je vždy konečný; produkty se ovšem tvoří v rozdílných množstvích. V některých případech je ovšem množství vznikajícího menšinového stereoizomeru příliš malé, než aby jej bylo možné zachytit pomocí dostupných analytických metod.
Enantioselektivní reakce je taková, při níž jeden enantiomer vzniká ve větším množství než jiný, nebo se vytváří opticky aktivní produkt z nechirálního výchozího materiálu za použití chirálního katalyzátoru, enzymu, nebo chirálního reaktantu. Míru selektivity udává enantiomerní přebytek.
Významnou variantou je kinetické rozlišení, při němž již existující chirální centrum reaguje s chirálním katalyzátorem, enzymem nebo chirálním katalyzátorem a jeden enantiomer reaguje rychleji než druhý a méně reaktivní enantiomer, nebo chirální centrum ovlivňuje reaktivitu reakčního centra v jiné části molekuly.
V diastereoselektivních reakcích vzniká jeden diastereomer přednostně před jiným (nebo část možných diastereomerů ve směsi produktů převažuje nad ostatními), čímž vzniká upřednostňovaná relativní stereochemie. Při této reakci se tvoří jen jedna stereochemie nebo část ze všech možných, případně již vytvořené chirální centrum (které nemusí být opticky čisté) selektivně vytváří jiné. Míru relativní selektivity udává diastereomerní přebytek.
Stereokonvergence je jev opačný ke stereospecificitě, spočívající v reakcích dvou nebo více stereoizomerů za vzniku produktu obsahujícího jediný stereoizomer.
Úroveň stereoselektivity se určuje výhradně podle produktů a jejich stereochemie.
Příklady
Příkladem stereoselektivní reakce je dehydrohalogenace 2-jodbutanu, při níž se z 60 % tvoří trans-but-2-en a z 20 % cisbut-2-en.[3] Jelikož jsou vzniklé alkeny geometrickými izomery a tedy také diastereomery, tak lze tuto reakci řadit mezi diastereoselektivní.

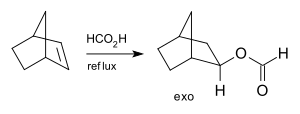
Adice kyseliny mravenčí na norbornen je rovněž stereoselektivní, protože vzniká pouze exo-izomer a nedochází k vytváření endo-izomeru:[4]
Pomocí Cramova pravidla lze předpovědět hlavní produkt diastereoselektivní nukleofilní adice na karbonylové skupiny sousedící s chirálními centry. Chirální centra nemusí být opticky čistá, protože relativní stereochemie bude u obou enantiomerů stejná. Jako příklad lze uvést níže zobrazenou reakci (S)-aldehydu s thiazolem vytvářející (S,S)-diastereomer a pouze malé množství (S,R)-diastereomeru:[5]

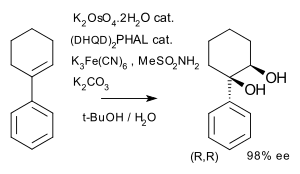
Sharplessova epoxidace je enantioselektivní reakce, při které se nechirální derivát allylalkoholu mění na opticky aktivní epoxyalkohol;při použití chirálnícjh allylalkoholů dochází ke kinetickému rozlišení. Další obdobnou reakci představuje Sharplessova asymetrická dihydroxylace. V níže zobrazeném případu z nechirálního alkenu vzniká pouze jeden ze čtyř možných stereoizomerů.[6]
Pokud se stereocentrum nachází vedle karbokationtu, tak může u mezi-[7] i vnitromolekulárních reakcí.[8][9] U níže zobrazené reakce se furanový nukleofil z nejméně stíněné strany přiblíží ke karbokationtu naproti terc-butylové skupině a výsledkem je vysoká diastereoselektivita:

Stereoselektivní biosyntéza
Biosyntézy pinoresinolu se účastní bílkoviny nazývané dirigentové proteiny; první byl objeven u zlatice prostřední (Forsythia intermedia). Tato bílkovina řídí stereoselektivní biosyntézu (+)-pinoresinolu z koniferylalkoholu.[10]
Druhý takový protein byl nalezen u huseníčku rolního (Arabidopsis thaliana), kde řídí enantioselektivní syntézu (−)-pinoresinolu.[11]
-Pinoresinol_Biosynthesis.svg.png.webp)
Odkazy
Související články
Reference
V tomto článku byl použit překlad textu z článku Stereoselectivity na anglické Wikipedii.
- (a)"Overlap Control of Carbanionoid Reactions. I. Stereoselectivity in Alkaline Epoxidation," Zimmerman, H. E.; Singer, L.; Thyagarajan, B. S. Journal of the American Chemical Society, 1959, 81, 108-116. (b)Eliel, E., "Stereochemistry of Carbon Compound", McGraw-Hill, 1962 pp 434-436.
- Například SN1 reakce zruší předchozí stereocentrum a poté vytvoří nové.
- Effects of base strength and size upon in base-promoted elimination reactions. Richard A. Bartsch, Gerald M. Pruss, Bruce A. Bushaw, Karl E. Wiegers Journal of the American Chemical Society; 1973; 95(10); 3405-3407. DOI:10.1021/ja00791a067
- Organic Syntheses Coll. Vol. 5, p.852 (1973); Vol. 42, p.79 (1962).
- Organic Syntheses, Coll. Vol. 10, p.140 (2004); Vol. 77, p.78 (2000)
- Organic Syntheses, Coll. Vol. 10, p.603 (2004); Vol. 79, p.93 (2002)
- Diastereoselective Friedel-Crafts Cyclization Reactions to 2-Substituted 1-Phenyl-1,2,3,4-tetrahydronaphthalenes Friedrich Mühlthau, Thorsten Bach Synthesis 2005: 3428-3436 DOI:10.1055/s-2005-918482
- High Facial Diastereoselectivity in Intra- and Intermolecular Reactions of Chiral Benzylic Cations Friedrich Mühlthau, Oliver Schuster, and Thorsten Bach Journal of the American Chemical Society, 2005, 127 (26), pp 9348–9349 DOI:10.1021/ja050626v
- Stereoselective Reactions with Stabilized Carbocations Pier Giorgio Cozzi and Fides Benfatti Angewandte Chemie International Edition 2009, 48 DOI:10.1002/anie.200905235
- L. B. Davin; H. B. Wang; A. L. Crowell. Stereoselective bimolecular phenoxy radical coupling by an auxiliary (dirigent) protein without an active center. Science. 1997, s. 362–366. DOI 10.1126/science.275.5298.362. PMID 8994027.
- B. Pickel; J. Pfannsteil; J. Conrad; U.Beifuss; A. Schaffer. An Enantiocomplementary Dirigent Protein for the Enantioselective Laccase-Catalyzed Oxidative Coupling of Phenols. Angewandte Chemie. 2007, s. 273–284. DOI 10.1007/s10086-007-0892-x.