Aktivace vazby uhlík-vodík
Aktivace vazby uhlík-vodík (C–H funkcionalizace) je druh chemické reakce, při níž se štěpí vazba mezi atomem uhlíku a vodíku a je nahrazena vazbou uhlík–X (kde X je obvykle uhlík, kyslík nebo dusík). Do štěpení vazby C-H bývá často zapojen atom přechodného kovu. Při tomto druhu reakce typicky reaguje uhlovodík s kovovým katalyzátorem za tvorby organokovového komplexu, ve kterém je uhlovodík koordinován na vnější sféru kovu. Meziprodukt vzniklý touto reakcí následně vstupuje do dalších reakcí, jež vytvářejí funkcionalizovaný produkt. Je podstatné, aby v průběhu funkcionalizace zůstává hydrokarbylový meziprodukt napojen na vnější sféru a je ovlivňován kovem.[1]
Někteří autoři označují pojmem C–H funkcionalizace všechny organické reakce, jejichž výsledky jsou přímé přeměny málo reaktivních vazeb C–H na vazby C–X, bez ohledu na mechanismus. Taková definice nevyžaduje koordinaci přechodného kovu na uhlovodík. Součástí této širší definice je i výše uvedená; ovšem také pod ni patří železem katalyzované funkcionalizace alkanů probíhající mechanismem s odrazem kyslíku (například u enzymů cytochromu P450 a jejich syntetických analogů), u kterých se do reakce pravděpodobně nezapojují vazby kov–uhlík. Patří sem ale na ligandech založené reakce mnoha karbenů kovů s uhlovodíky, i když některé mohou probíhat více mechanismy. Aktivace vazby C–H se někdy také definuje jako štěpení vazeb C–H jakýmkoliv mechanismem vedoucím k funkcionalizaci uhlovodíkové skupiny. Též se objevuje používání pojmu C–H aktivace u užší definice, zatímco se v širší definici používá název C–H funkcionalizace.
Rozdělení
Mechanismy C-H aktivací lze rozdělit na tři základní druhy:
- (i) Oxidační adice, kde se kovové centrum s nízkým oxidačním číslem váže na vazbu mezi uhlíkem a vodíkem, čímž ji odštěpí a kov se zoxiduje.
- LnM + RH → LnMR(H)
- (ii) Elektrofilní aktivace, během nichž elektrofilní kov atakuje uhlovodík a odštěpuje proton:
- LnM+ + RH → LnMR + H+
substituent se na substrát naváže elektrofilní aromatickou substitucí.
- (iii) Metateze vazby sigma, probíhající přes „čtyřcentrovaný“ meziprodukt, jehož vazby se rozštěpí a vytvoří konečný produkt:
- LnMR + R'H → LnMR' + RH
Historie
První aktivace vazby C-H se často připisuje Ottu Dimrothovi, který v roce 1902 oznámil, že benzen reaguje s octanem rtuťnatým. Této reakce, podobné Friedelovým–Craftsovým reakcím, se může účastnit mnoho různých elektrofilních kovů. Joseph Chatt provedl adici vazeb C-H u naftalenu na komplexy ruthenia.[2]
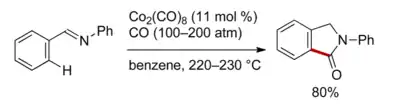
Aktivace vazeb uhlík-vodík mohou být provedeny s využitím chelatace; příkladem je kobaltem katalyzovaná chelatací asistovaná C-H funkcionalizace 2-fenylisoindolin-1-onu (E)-N,1-difenylmethaniminem.[3]
V roce 1969 byla popsána reakce methanu a těžké vody za přítomnosti tetrachloroplatnatanu draselného. Navržený mechanismus zahrnuje navázání methanu na platnatou sloučeninu. Roku 1972 popsala stejná skupina přípravu methanolu a chlormethanu obdobnou reakcí methanu a vody s použitím stechiometrického množství tetrachloroplatnatanu draselného a katalytického množství hexachloroplatiničitanu draselného. Tento postup je jedním z mála známých plně katalytických způsobů funkcionalizace alkanů.[1][4]
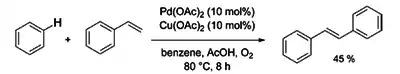
V roce 1969 oznámil Yuzo Fudžiwara přípravu (E)-1,2-difenylethenu z benzenu a styrenu pomocí Pd(OAc)2 a Cu(OAc)2.[5]
M. L. H. Green roku 1970 objevil fotochemickou reakci spočívající v navázání wolframu (ve formě komplexu Cp2WH2) na vazby C–H u benzenu[6] a George M. Whitesides v roce 1979 provedl jako první vnitromolekulární alifatickou C–H aktivaci.[7]
Dalšího pokroku dosáhly nezávisle na sobě dvě skupiny v roce 1982. Robert G. Bergman popsal přechodným kovem řízenou mezimolekulární aktivaci vazby uhlík-vodík u neaktivovaných a zcela nenasycených uhlovodíků pomocí oxidační adice. Fotolýzou Cp*Ir(PMe3)H2 (Cp* = pentamethylcyklopentadienyl) připravil koordinačně nenasycenou sloučeninou Cp*Ir(PMe3), která reagovala v oxidační adici s cyklohexanem a neopentanem za tvorby příslušných hydridoalkylových komplexů, Cp*Ir(PMe3)HR, (R = cyklohexyl či neopentyl).[8]
W. A. G. Graham zjistil, že stejné uhlovodíky reagují s Cp*Ir(CO)2 po vystavení světlu za vzniku alkylhydridokomplexů Cp*Ir(CO)HR, kde R je cyklohexyl či neopentyl.[9]
U této reakce se předpokládá, že probíhá přes oxidační adici alkanu na šestnáctielektronový iridný meziprodukt Cp*Ir(CO), jenž se fotochemicky vytváří z Cp*Ir(CO)2.
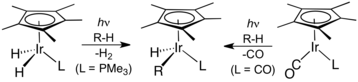 Aktivace vazby C–H podle Bergmana et al. (vlevo) a Grahama et al.
Aktivace vazby C–H podle Bergmana et al. (vlevo) a Grahama et al.
Byly popsány selektivní aktivace a funkcionalizace alkanových vazeb C–H pomocí komplexů wolframu s pentamethylcyklopentadienylovými, nitrosylovými, allylovými a neopentylovými ligandy, Cp*W(NO)(η3-allyl)(CH2CMe3).[10]
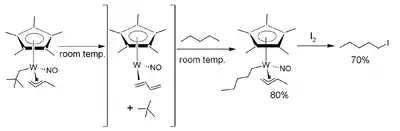 Aktivace vazby C–H u pentanu
Aktivace vazby C–H u pentanu
Jedním z příkladů využití tohoto postupu je selektivní přeměna pentanu na halogenovaný uhlovodík 1-jodpentan. Provádí se termolýzou Cp*W(NO)(η3-allyl)(CH2CMe3) v pentanu za pokojové teploty, vedoucí k eliminaci neopentanu v reakci reakci pseudoprvního řádu, přes nedetekovatelný elektronově a stericky nenasycený 16elektronový meziprodukt koordinovaný s η2-buta-1,3-dienovým ligandem. Následnou mezimolekulární aktivací pentanu (sloužícího jako rozpouštědlo) vzniká 18elektronový komplex obsahující n-pentylový ligand. V jiném kroku reakcí s jodem při −60 °C se z komplexu uvolní 1-jodpentan.
Řízená C-H aktivace
Pod řízenou C-H aktivaci spadají postupy, které využívají řídicí skupiny ovlivňující regiochemii a stereochemii.[11]
Tento postup je nejúčinnějším známým postupem aktivace vazeb C-H. N,N-dimethylbenzylamin může být snadno cyklometalován řadou přechodných kovů.[12]
Bylo zavedeno několik úprav, jako je například použití slabě koordinujících skupin v Muraiově reakci.[13]

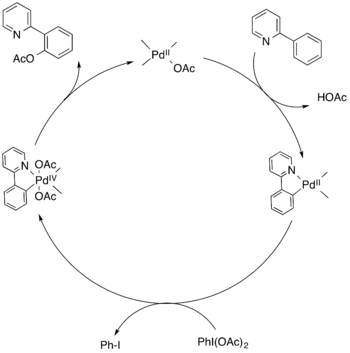
Mechanismus C-H aktivace 2-fenylpyridinu katalyzované palladiem zahrnuje metalocyklický meziprodukt. Tento meziprodukt se oxiduje na palladičitou sloučeninu, u níž poté proběhne redukční eliminace vazby C-O a vytvoří se konečný produkt.[14]
Borylace
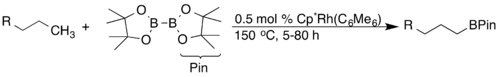
Přeměna vazeb C-H na vazby C-B skrze borylační reakce byla díky své syntetické využitelnosti prozkoumána podrobně. John F. Hartwig popsal vysoce regioselektivní borylace alkanů a arenů katalyzované komplexy rhodia. U alkanů docházelo výhradně ke koncové funkcionalizaci.[15]
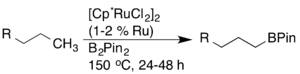
Později bylo zjištěno, že použitím katalyzátorů založených na rutheniu lze dosáhnout vyšší aktivity a kompatibility funkčních skupin.[16]
Byly vyvinuty i jiné katalyzátory borylací, například založené na iridiu, dobře využitelné pro aktivace vazeb C-H.[17][18][19]
U zemního plynu
Přírodní methan se i přes svou nízkou cenu a snadnou dostupnost příliš nepoužívá v chemickém průmslu. Jeho hlavní využití je při výrobě syntézního plynu, což je směs oxidu uhelnatého a vodíku. Tato směs se následně používá při Fischerově–Tropschově syntéze uhlovodíků s delšími řetězci či methanolu.[20][21]
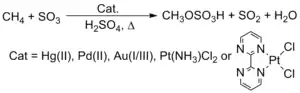
U takto získaných uhlovodíků lze provést aktivaci vazeb C-H. Periana zjistil, že komplexy některých přechodných kovů, jako jsou Pt, Pd, Au a Hg, reagují s methanem (CH4) v kyselině sírové (H2SO4) za vzniku methylmonosulfátu.[22][23]
Asymetrické C-H aktivace
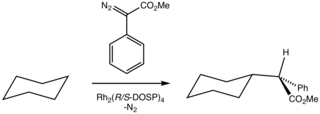
Součástí totální syntézy kyseliny lithospermiové je C-H funkcionalizace vedoucí ke vzniku vysoce funkcionalizovaného systému. Řídicí skupinou je zde chirální neracemický imin, který provádí vnitromolekulární alkylaci, jež umožňuje rhodiem katalyzovanou přeměnu iminu na dihydrobenzofuran.[25]

Totální syntéza kalothrixinu A a B zahrnuje vnitromolekulární palladiem katalyzovanou řízenou C-H aktivaci. Dochází zde k reakci vazeb C-I a C-H za tvorby vazby C-C bond.[26]

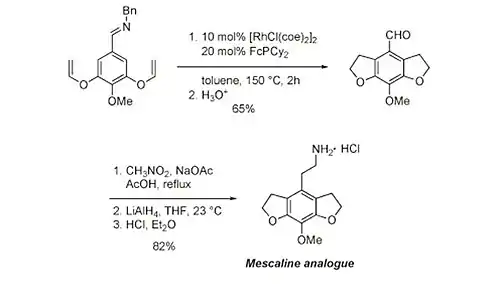
Analog meskalinu se připravuje rhodiem katalyzovanou enantioselektivní reakcí aryliminu zahrnující C-H aktivaci.[27]
Reference
V tomto článku byl použit překlad textu z článku Carbon–hydrogen bond activation na anglické Wikipedii.
- Parthasarathy Gandeepan; Thomas Müller; Daniel Zell; Gianpiero Cera; Svenja Warratz; Lutz Ackermann. 3d Transition Metals for C–H Activation. Chemical Reviews. 2019, s. 2192–2452. DOI 10.1021/acs.chemrev.8b00507. PMID 30480438.
- J. Chatt; J. M. Davidson. The tautomerism of arene and ditertiary phosphine complexes of ruthenium(0) and the preparation of new types of hydrido-complexes of ruthenium(II). Journal of the Chemical Society. 1965, s. 843. DOI 10.1039/JR9650000843.
- Shunsuke Murahashi. Synthesis of Phthalimidines from Schiff Bases and Carbon Monoxide. Journal of the American Chemical Society. 1955-12-01, s. 6403–6404. ISSN 0002-7863. DOI 10.1021/ja01628a120.
- U. Fekl; K. I. Goldberg. Homogeneous Hydrocarbon C-H Bond Activation and Functionalization with Platinum. [s.l.]: [s.n.] ISBN 9780120236541. DOI 10.1016/S0898-8838(03)54005-3. S. 259–320.
- Yuzo Fujiwara; Ichiro Noritani; Sadao Danno; Asano Ryuzo; Shiichiro Teranishi. Aromatic substitution of olefins. VI. Arylation of olefins with palladium(II) acetate. Journal of the American Chemical Society. 1969-12-01, s. 7166–7169. ISSN 0002-7863. DOI 10.1021/ja01053a047. PMID 27462934.
- M. L. Green; P. J. Knowles. Formation of a tungsten phenyl hydride derivatives from benzene. Journal of the Chemical Society D. 1970, s. 1677. DOI 10.1039/C29700001677.
- Paul Foley; George M. Whitesides. Thermal generation of bis(triethylphosphine)-3,3-dimethylplatinacyclobutane from dineopentylbis(triethylphosphine)platinum(II). Journal of the American Chemical Society. 1979, s. 2732–2733. DOI 10.1021/ja00504a041.
- Andrew H. Janowicz; Robert G. Bergman. Carbon–hydrogen activation in saturated hydrocarbons: direct observation of M + R−H → M(R)(H). Journal of the American Chemical Society. 1982, s. 352–354. DOI 10.1021/ja00365a091.
- James K. Hoyano; A. G. William. Oxidative addition of the carbon–hydrogen bonds of neopentane and cyclohexane to a photochemically generated iridium(I) complex. Journal of the American Chemical Society. 1982, s. 3723–3725. DOI 10.1021/ja00377a032.
- Rhett A. Baillie; Peter Legzdins. Distinctive Activation and Functionalization of Hydrocarbon C–H Bonds Initiated by Cp*W(NO)(η3-allyl)(CH2CMe3) Complexes. Accounts of Chemical Research. 2013, s. 330–340. DOI 10.1021/ar400108p. PMID 24047442.
- T. Brückl; R. D. Baxter; Y. Ishihara; P. S. Baran. Innate and Guided C-H Functionalization Logic. Accounts of Chemical Research. 2012, s. 826–839. DOI 10.1021/ar200194b. PMID 22017496.
- Michael J. Chetcuti; Vincent Ritleng. Formation of a Ruthenium–Arene Complex, Cyclometallation with a Substituted Benzylamine, and Insertion of an Alkyne. Journal of Chemical Education. 2007, s. 1014. DOI 10.1021/ed084p1014. Bibcode 2007JChEd..84.1014C.
- Shinji Murai; Fumitoshi Kakiuchi; Shinya Sekine; Yasuo Tanaka; Asayuki Kamatani; Motohiro Sonoda; Naoto Chatani. Efficient catalytic addition of aromatic carbon–hydrogen bonds to olefins. Nature. 1993, s. 529–531. DOI 10.1038/366529a0. Bibcode 1993Natur.366..529M.
- T. W. Lyons; M. S. Sanford. Palladium-Catalyzed Ligand-Directed C–H Functionalization Reactions. Chemical Reviews. 2010, s. 1147–1169. DOI 10.1021/cr900184e. PMID 20078038.
- Huiyuan Chen; Sabine Schlecht; Thomas C. Semple; John F. Hartwig. Thermal, Catalytic, Regiospecific Functionalization of Alkanes. Science. 2000, s. 1995–1997. DOI 10.1126/science.287.5460.1995. PMID 10720320. Bibcode 2000Sci...287.1995C.
- J. M. Murphy; J. D. Lawrence; K. Kawamura; C. Incarvito; J. F. Hartwig. Ruthenium-Catalyzed Regiospecific Borylation of Methyl C-H bonds. Journal of the American Chemical Society. 2006, s. 13684–13685. DOI 10.1021/ja064092p. PMID 17044685.
- T. Ishiyama; J. Takagi; K. Ishida; N. Miyaura; N. R. Anastasi; J. F. Hartwig. Mild Iridium-Catalyzed Borylation of Arenes. High Turnover Numbers, Room Temperature Reactions, and Isolation of a Potential Intermediate. Journal of the American Chemical Society. 2002, s. 390–391. DOI 10.1021/ja0173019. PMID 11792205.
- T. Ishiyama; J. Takagi; J. F. Hartwig; N. Miyaura. A Stoichiometric Aromatic C-H Borylation Catalyzed by Iridium(I)/2,2′-Bipyridine Complexes at Room Temperature. Angewandte Chemie International Edition. 2002, s. 3056–3058. DOI 10.1002/1521-3773(20020816)41:16<3056::aid-anie3056>3.0.co;2-#.
- L. P. Press; A. J. Kosanovich; B. J. McCulloch; O. V. Ozerov. High-Turnover Aromatic C–H Borylation Catalyzed by POCOP-Type Pincer Complexes of Iridium. Journal of the American Chemical Society. 2016, s. 9487–9497. DOI 10.1021/jacs.6b03656. PMID 27327895.
- A. Sen. Activation of Unreactive Bonds and Organic Synthesis. [s.l.]: Springer Berlin Heidelberg, 1999. ISBN 978-3-540-64862-8. Kapitola Catalytic Activation of Methane and Ethane by Metal Compounds, s. 81–95.
- Methanol [online]. [cit. 2016-02-01]. Dostupné online.
- R. A. Periana; D. J. Taube; E. R. Evitt; D. G. Loffler; P. R. Wentrcek; G. Voss; T. Masuda. A Mercury-Catalyzed, High-Yield System for the Oxidation of Methane to Methanol. Science. 1993, s. 340–343. DOI 10.1126/science.259.5093.340. Bibcode 1993Sci...259..340P.
- R. A. Periana; D. J. Taube; S. Gamble; H. Taube; T. Satoh; H. Fujii. Platinum Catalysts for the High-Yield Oxidation of Methane to a Methanol Derivative. Science. 1998, s. 560–564. DOI 10.1126/science.280.5363.560. PMID 9554841. Bibcode 1998Sci...280..560P.
- H. M. L. Davies; D. Morton. Guiding Principles for Site Selective and Stereoselective Intermolecular C–H Functionalization by Donor/Acceptor Rhodium Carbenes. Chemical Society Reviews. 2011, s. 1857–1869. DOI 10.1039/C0CS00217H. PMID 21359404.
- S. J. O'Malley; K. L. Tan; A. Watzke; R. G. Bergman; J. A. Ellman. Total Synthesis of (+)-Lithospermic Acid by Asymmetric Intramolecular Alkylation via Catalytic C-H Bond Activation. Journal of the American Chemical Society. 2005, s. 13496–13497. DOI 10.1021/ja052680h. PMID 16190703.
- N. Ramkumar; R. Nagarajan. b. Total Synthesis of Calothrixin A and B via C-H Activation. The Journal of Organic Chemistry. 2013, s. 2802–2807. DOI 10.1021/jo302821v. PMID 23421392.
- Ahrendt Kateri A.; Robert G. Bergman; Jonathan A. Ellman. Synthesis of a Tricyclic Mescaline Analogue by Catalytic C−H Bond Activation. Organic Letters. 2003-04-01, s. 1301–1303. DOI 10.1021/ol034228d. PMID 12688744.