Hydroformylace
Hydroformylace je organická reakce používaná na výrobu aldehydů z alkenů.[1][2] Jde o adici formylové skupiny a atomu vodíku na dvojnou vazbu mezi atomy uhlíku. Tento proces má velký význam, protože aldehydy lze snadno přeměnit na další látky, například hydrogenací na alkoholy, ze kterých se následně dají vyrobit detergenty. Využívá se také na výrobu sloučenin používaných při syntéze vůní a léčiv.
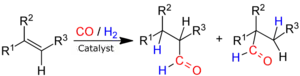
Reakce se obvykle provádí za vysokého tlaku (1 až 10 MPa) a teplot od 40 do 200 °C, kdy reaguje alken s oxidem uhelnatým a vodíkem. [3] Používají se katalyzátory obsahující přechodné kovy, které se v reakční směsi rozpouští, a tak hydroformylace patří mezi homogenní katalytické procesy.
Historie
Hydroformylaci objevil německý chemik Otto Roelen v roce 1938 při zkoumání Fischerovy–Tropschovy syntézy, když v F-T reaktoru smíchal diethylketon s aldehydy. Během svého výzkumu objevil užitečnost katalyzátorů založených na kobaltu.Tetrakarbonylhydrokobalt, který byl objeven a izolován o několik let dříve, se ukázal jako velmi dobrý katalyzátor hydroformylace.[4][5] Později se ukázalo, že tributylfosfin (PBu3) zvyšuje selektivitu hydroformylace katalyzované kobaltem. Mechanismus hydroformylace vysvětlili Richard F. Heck a David S. Breslow v 60. letech 20. století.[6]
V roce 1968 byly popsány vysoce aktivní katalyzátory obsahující rhodium.[7] Od 70. let 20. století se většina hydroformylací provádí s těmito katalyzátory.[8] Jsou známy i katalyzátory rozpustné ve vodě, které usnadňují oddělení produktů od katalyzátoru.[9]
Mechanismus
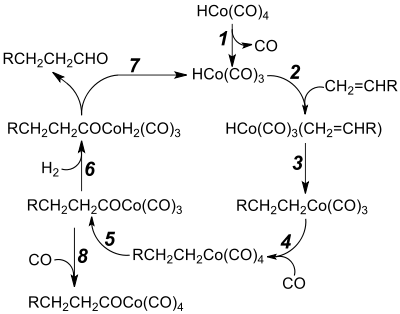
Hydroformylace katalyzované kobaltem začínají odštěpením oxidu uhelnatého z tetrakarbonylhydridu kobaltu za vzniku 16elektronového meziproduktu (krok 1). Následným navázáním alkenu vznikne 18elektronová molekula (krok 2). V kroku 3 dochází k přeměně na 16elektronový alkyltrikarbonyl. Koordinací další molekuly CO vznikne alkyltetrakarbonyl (krok 4).[6] Přesunem CO se utvoří 16elektronový acyl (5). V následujícím kroku (6) se oxidační adicí vodíku vytvoří dihydridokomplex, jenž poté (7) redukční eliminací uvolní aldehyd.[10] Od 70. let 20. století se většina hydroformylací provádí s těmito katalyzátory. Krok 8 je neproduktivní a vratný.
Selektivita
Důležitým parametrem hydroformylace je „normální“/„iso“ selektivita; například při hydroformylaci propenu mohou vznikat dva různé produkty, butanal a isobutanal:
- H2 + CO + CH3CH=CH2 → CH3CH2CH2CHO („normální“)
- vs.
- H2 + CO + CH3CH=CH2 → (CH3)2CHCHO („iso“)
Vznik jednoho či druhého izomeru je v souladu s regiochemií navázání alkenu na vazbu M-H. Protože je „normální“ produkt stabilnější, a tvoří se tak ve větším množství, tak se výzkum většinou soustředí na nalezení vhodného katalyzátoru pro tento izomer.
Vliv sterických efektů
Adice hydridu kobaltu na primární alkeny podle Markovnikovova pravidla je kvůli sterickým efektům mezi kobaltovým centrem a sekundárním alkylovým ligandem energeticky nevýhodná. Při použití smíšených karbonylo/fosfinových komplexů se často dosahuje větší selektivity takové adice a tvoří se tak převážně produkty s nerozvětvenými řetězci (n-aldehydy). Součástí moderních katalyzátorů bývají chelatující ligandy, například difosfity.[11]
Vliv elektronových efektů
Čím větší je elektronová hustota na hydridovém komplexu, tím méně jsou vlastnosti tohoto komplexu podobné protonu. Elektronové efekty, které obvykle podporují Markovnikovovu adici na alken, tak mají na reakci menší vliv, což vede k tomu, že hydridy s vyšší elektronovou hustotou reagují selektivněji.
Tvorba acylu
K omezení izomerizace alkenu, která je vedlejší reakcí, je třeba urychlit reakcí karbonylu s organokovovým centrem. Rychlost připojování karbonylového uhlíku na vazbu C-M se zvyšuje s rychlostí beta-hydridové eliminace.[12]
Asymetrická hydroformylace
Hydroformylací prochirálních alkenů se vytvářejí nová stereocentra. Při použití chirálních fosfinových ligandů se bude jeden enantiomer vytvářet ve větším množství než druhý.[13] Například dexibuprofen, (+)-(S)-enantiomer ibuprofenu, lze připravit enantioselektivní hydroformylací a následnou oxidací.
Využití v průmyslu
Průmyslové procesy využívající hydroformylaci mohou být značně odlišné v závislosti na délce řetězce hydroformylovaného alkenu, použitém katalyzátoru (kovu i ligandu) a způsobu obnovy katalyzátoru. V původním procesu Ruhrchemie se vyráběl propanal z ethenu a syntézního plynu za katalýzy hydridem tetrakarbonylu kobaltu. V současnosti se postupy využívající katalyzátory založené na kobaltu používají u alkenů se středně dlouhými až dlouhými řetězci, zatímco při hydroformylaci propenu se používají katalyzátory s obsahem rhodia, které jsou mnohem nákladnější; při hydroformylaci alkenů s dlouhými řetězci se obtížně odděluje katalyzátor od produktu.
BASF oxo proces
BASF oxo procesem se hydroformylují převážně alkeny s dlouhými řetězci, používají se přitom katalyzátory obsahující kobalt.[14] Reakce se provádí za nižších teplot, přičemž se tvoří převážně nerozvětvené produkty. Používá se tlak kolem 30 MPa a teplota 150 až 170 °C. Katalyzátor se z kapalného produktu obnovuje oxidací na kobaltnatou sloučeninu, která je rozpustná ve vodě, a následným přidáním vodného roztoku kyseliny mravenčí nebo octové. Tímto způsobem se katalyzátor dostane do roztoku, ze kterého může být obnoven. Ztráty se vyrovnávají přídavkem kobaltnatých solí.[15]
Exxon proces
Exxon proces, také známý jako Kuhlmannův oxo proces, se používá k hydroformylaci alkenů s 6 až 12 atomy uhlíku, používají se katalyzátory založené na kobaltu. Katalyzátor se obnovuje přidáním vodného roztoku hydroxidu nebo uhličitanu sodného do reakční směsi. Extrakcí s alkenem a neutralizací kyselinou sírovou se obnoví karbonylhydrid kovu, při tom se používá syntézní plyn, který je absorbován alkenem a vrací se do reaktoru. Podobně jako u BASF procesu reakce probíhá při tlaku 30 MPa a teplotě 160 až 180 °C.[15]
Shell proces
Při Shell procesu se k hydroformylaci používají kobaltové katalyzátory pozměněné fosfinovými ligandy, rektanty jsou alkeny se 7 až 14 atomy uhlíku. Vzniklé aldehydy se následně hydrogenují na mastné alkoholy, které se oddělují destilací, díky čemuž je možné obnovit katalyzátor. Mezi produkty převažují ty s nerozvětvenými řetězci, ze kterých se dále vyrábějí detergenty. Tlak při reakci bývá 4 až 8 MPa a teplota 150 až 190 °C.[15]
Union Carbide proces
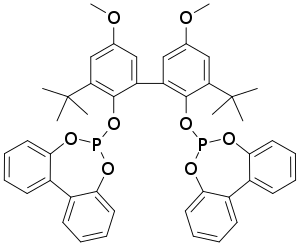
Při Union Carbide (UCC) procesu se používají katalyzátory obsahující rhodium, rozpuštěné ve vysokovroucím oleji, vznikají přitom hydroformylací propenu produkty s vyššími molárními hmotnostmi. Po reakci se oddělí těkavé složky reakční směsi a zbytek se destiluje, čímž dojde k izolaci butyraldehydu, zatímco katalyzátor s vedlejšími produkty se vrací do reaktoru. Tlak při reakci bývá přibližně 1,8 MPa a teplota 95 až 100 °C.[15] Součástí katalyzátoru bývá difosfitový ligand BiPhePhos.[16][17]
Ruhrchemie/Rhône-Poulenc proces
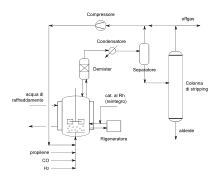
Při Ruhrchemie/Rhône-Poulenc procesu se používá katalyzátor založený na rhodiu, na které je navázána trisodná sůl kyseliny 3,3′,3″-fosfantriyltris(benzensulfonové) (TPPTS).[18] Trisulfonace trifenylfosfinového ligandu dává komplexu hydrofilní vlastnosti. Katalytický komplex obsahuje devět sulfonátových skupin a velmi dobře rozpustný ve vodě (kolem 1 kg/l), ovšem nerozpouští se ve fázi, v níž vzniká produkt.[19] TPPTS se používá v asi 50násobném přebytku, čímž se zabraňuje vyluhování katalyzátoru. Jako reaktanty se používají propen a syntézní plyn s poměrem vodíku a oxidu uhelnatého 1,1:1. Vzniká směs butyraldehydu a isobutyraldehydu v poměru 96:4 s několika vedlejšími produkty, jako jsou alkoholy a estery.[19] Ruhrchemie/Rhône-Poulenc proces se stal první průmyslově využívanou dvoufázovou metodou, ve které byl použit katalyzátor ve vodném roztoku.
V průběhu reakce se vytváří organická fáze, která se postupně odděluje, zatímco vodná fáze zůstává v reaktoru.[19]
Reakce se provádí za promíchávání reakční směsi v reaktoru, přičemž se syntézní plyn a alken dodávají zespodu do katalyzátorové fáze za neustálého silného míchání. Vznikající aldehydová fáze se v horní části reaktoru odděluje od vodné fáze. Vodný roztok obsahující katalyzátor se opětovně zahřívá pomocí výměníku tepla a vrací se do reaktoru.[19] Přebytek alkenu a syntézního plynu se odděluje od aldehydové fáze a také vrací do reaktoru. Teplo uvolněné při reakci se využívá k tvorbě páry, která slouží k destilaci organické fáze a oddělení butyraldehydu od isobutyraldehydu.[20] Do organické fáze se mohou dostat katalyzátorové jedy, které se odstraňují reakcemi s aldehydem, a tím nedochází k jejich hromadění, díky čemuž není nutné syntézní plyn přečišťovat.
Nealkenové substráty
Ligandy obsahující rhodium či oktakarbonyl dikobaltu katalyzují hydroformylace formaldehydu na glykolaldehyd a ethylenoxidu na 3-hydroxypropanal. Tyto produkty lze dále hydrogenovat na ethan-1,2-diol nebo propan-1,3-diol. Reakce nejlépe probíhá v zásaditých rozpouštědlech (například pyridinu).[21][22]
Při použití oktakarbonylu dikobaltu (Co2(CO)8) jako katalyzátoru může za nepřítomnosti vodíku reakcí ethenu s oxidem uhelnatým vznikat pentan-3-on. Meziproduktem je zde [CH3C(O)Co(CO)3(ethen)], z něhož se následně vytvoří [CH3COCH2CH2Co(CO)3]. Vodík potřebný pro tuto reakci pochází z reakce vodní páry s oxidem uhelnatým.[23]
Pokud reakce H2O a CO neproběhne, tak se vytvoří polymer, ve kterém se střídají monomery oxidu uhelnatého a ethylenové skupiny. Takové alifatické polyketony se lépe připravují za použití katalyzátorů založených na palladiu.[24]
Funkcionalizované alkeny, jako je allylalkohol, lze také hydroformylovat. Z allylalkoholu vzniká butan-1,4-diol, ze kterého lze provést izomerizaci za přítomnosti katalyzátoru, například komplexu trifenylfosfinu s rhodiem. Při použití komplexů kobaltu by vznikal propanal.[25] Hydroformylace alkenylesterů a alkenyletherů probíhá obvykle v poloze α vůči esterové/etherové skupině.
Hydroformylací kyseliny akrylové a methakrylové vzniká v prvním kroku Markovnikovův produkt.[20] Změnou podmínek reakce lze vytvořit různé produkty. Při vysokých teplotách a nízkém tlkaku oxidu uhelnatého dojde k izomerizaci Markovnikovova produktu na stabilnější β izomer, ze kterého se vytvoří n-aldehyd. Nízká teplota a vysoký tlak CO za přebytku fosfinu, blokujícího koordinační místa, způsobí, že hydroformylace bude přednostně probíhat v poloze α a k izomerizaci nedojde.[20]
Vedlejší a následné reakce
U alkenů
K vedlejším reakcím alkenů patří izomerizace a hydrogenace na dvojné vazbě. Alkany vytvořené touto hydrogenací se hydroformylace neúčastní, ovšem izomerizace s následnou tvorbou n-alkylových komplexů je žádoucí reakcí. Hydrogenace obvykle probíhá v menší míře, ovšem při použití kobaltových katalyzátorů s fosfiny se může hydrogenovat až 15 % alkenu.
U aldehydů
Častou žádoucí reakcí aldehydů je jejich hydrogenace na alkoholy. Při vyšších teplotách a parciálních tlacích vodíku je pravděpodobnost takové reakce vyšší. Aldehyd nejspíše na začátku vytvoří CO-π-komplex s katalyzátorem, následně dojde k přesmyku na alkoxidový komplex a poté se oxidační adicí odštěpí vodík a vznikne alkohol a vytvoří se komplex, který reaguje na začátku hydroformylace.
Vazba uhlík-kyslík v molekule aldehydu se také může účastnit hydroformylace, přičemž vznikají kyselina mravenčí a její estery. K této reakci je nutné navázání oxdu uhelnatého na vazbu kyslík-kov v alkoxidovém komplexu. Vzniklý formylový komplex se následně přemění oxidační adicí vodíku na ester kyseliny mravenčí. Aldehydy pak mohou dále reagovat aldolovou kondenzací na prekurzory požadovaných produktů, jako je 2-ethylhexenal, nebo vysokomekulární produkty.
U katalyzátoru a ligandů
Účinnost fosfinových ligandů závisí na reakčních podmínkách. Trifenylfosfin podléhá hydrogenolýze za vzniku benzenu a difenylfosfinu. Navázání oxidu uhelnatého na vazbu kov-fenyl v meziproduktu může vést k tvorbě benzaldehydu, který může poté být hydrogenován na benzylalkohol.[26] Jedna z fenylových skupin ligandu může být nahrazena propenen a vzniklý difenylpropylfosfin tak může inhibovat hydroformylaci kvůli své zvýšené zásaditosti.[26]
Reference
V tomto článku byl použit překlad textu z článku Hydroformylation na anglické Wikipedii.
- Robert Franke; Detlef Selent; Armin Börner. Applied Hydroformylation. Chemical Reviews. 2012, s. 5675–5732.
- T. Ojima; C.-Y. Tsai; M. Tzamarioudaki; D. Bonafoux. The Hydroformylation Reaction. Organic Reactions. 2000, s. 1.
- P. Pino; C. Botteghi. Aldehydes from olefins: cyclohexanecarboxaldehyde. Organic Syntheses. 1977, s. 11.
- Boy Cornils; Wolfgang A. Herrmann; Manfred Rasch. Otto Roelen, Pioneer in Industrial Homogeneous Catalysis. Angewandte Chemie International Edition in English. 1994, s. 2144–2163.
- Archived copy [online]. [cit. 2007-01-07]. Dostupné v archivu pořízeném z originálu dne 2007-09-28.
- Richard F. Heck; David S. Breslow. The Reaction of Cobalt Hydrotetracarbonyl with Olefins. Journal of the American Chemical Society. 1961, s. 4023–4027.
- D. Evans; J. A. Osborn; G. Wilkinson. Hydroformylation of Alkenes by Use of Rhodium Complex Catalyst. Journal of the Chemical Society. 1968, s. 3133–3142.
- J. F. Hartwig; Organotransition metal chemistry – from bonding to catalysis. University Science Books. 2009. 753, 757–578. ISBN 978-1-891389-53-5
- Cornils, B.; Herrmann, W. A. (eds.) “Aqueous-Phase Organometallic Catalysis” VCH, Weinheim: 1998
- Jack Halper. Organometallic chemistry at the threshold of a new millennium. Retrospect and prospect. Pure and Applied Chemistry. 2001, s. 209–220.
- Aitor Gual; Cyril Godard; Verónica de la Fuente; Sergio Castillón. Phosphorus(III) Ligands in Homogeneous Catalysis: Design and Synthesis. [s.l.]: [s.n.], 2012. ISBN 9781118299715. Kapitola Design and Synthesis of Phosphite Ligands for Homogeneous Catalysis, s. 81–131.
- M. Kuil; T. Soltner; P. W. N. van Leeuwen; J. N. H. Reek. High-Precision Catalysts: Regioselective Hydroformylation of Internal Alkenes by Encapsulated Rhodium Complexes. Journal of the American Chemical Society. 2006, s. 11344–11345.
- Gene W. Wong; Tyler T. Adint; Clark R. Landis. Synthesis of (2R)-3-[[(1,1-Dimethylethyl)dimethylsilyl]oxy]-2-methylpropanal by Rhodium-Catalyzed Asymmetric Hydroformylation. Organic Syntheses. 2012, s. 243.
- G. Duembgen; D. Neubauer. Grosstechnische Herstellung von Oxo-Alkoholen aus Propylen in der BASF. Chemie Ingenieur Technik – CIT. 1969, s. 974–980.
- Boy Cornils, Wolfgang A. Herrmann, Chi-Huey Wong, Horst Werner Zanthoff: Catalysis from A to Z: A Concise Encyclopedia, 2408 Seiten, Verlag Wiley-VCH Verlag GmbH & Co. KGaA, (2012), ISBN 3-527-33307-X.
- Gregory D. Cuny; Stephen L. Buchwald. Practical, High-Yield, Regioselective, Rhodium-Catalyzed Hydroformylation of Functionalized α-olefins. Journal of the American Chemical Society. 1993, s. 2066–2068.
- Annemiek Van Rooy; Paul C. J. Kamer; Piet W. N. M. Van Leeuwen; Kees Goubitz; Jan Fraanje; Nora Veldman; Anthony L. Spek. Bulky Diphosphite-Modified Rhodium Catalysts: Hydroformylation and Characterization. Organometallics. 1996, s. 835–847. Dostupné online.
- W. A. Herrmann, C. W. Kohlpaintner, Angewandte Chemie 1993, 105, 1588
- Ernst Wiebus; Boy Cornils. Die großtechnische Oxosynthese mit immobilisiertem Katalysator. Chemie Ingenieur Technik. 1994, s. 916–923.
- Jürgen Falbe, Ch. R. Adams: Carbon Monoxide in Organic Synthesis, Springer Verlag, 1970, ISBN 3-540-04814-6
- A. S. C. Chan; H-S. Shieh. A mechanistic study of the homogeneous catalytic hydroformylation of formaldehyde: synthesis and characterization of model intermediates. Inorganica Chimica Acta. 1994, s. 89–95.
- A. Spencer. Hydroformylation of formaldehyde catalysed by rhodium complexes. Journal of Organometallic Chemistry. 1980, s. 113–123.
- K. Murata; A. Matsuda. Application of Homogeneous Water-Gas Shift Reaction III Further Study of the Hydrocarbonylation – A highly Selective Formation of Diethyl Keton from Ethene, CO and H2O. Bulletin of the Chemical Society of Japan. 1981, s. 2089–2092.
- J. Liu; B. T. Heaton; J. A. Iggo; R. Whyman. The Complete Delineation of the Initiation, Propagation, and Termination Steps of the Carbomethoxy Cycle for the Carboalkoxylation of Ethene by Pd–Diphosphane Catalysts. Angewandte Chemie International Edition. 2004, s. 90–94.
- Bernhard Fell; Wolfgang Rupilius; Friedrich Asinger. Zur Frage der Isomerenbildung bei der Hydroformylierung höhermolekularer Olefine mit komplexen Kobalt- und Rhodiumkatalysatoren. Tetrahedron Letters. 1968, s. 3261–3266.
- Arno Behr: Angewandte homogene Katalyse, Wiley-VCH. Weinheim, ISBN 3-527-31666-3