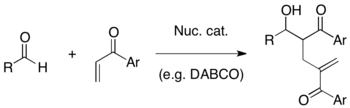
Baylisova–Hillmanova reakce
Baylisova–Hillmanova reakce je organická reakce vytvářející vazby uhlík–uhlík na pozicích α aktivovaných alkenů reakcemi s uhlíkatými elektrofily, jako jsou aldehydy. Katalyzátorem bývá nukleofil, například terciární amin nebo fosfin, a produkt mívá vysokou míru funkcionalizace (funkcionalizovaný allylalkohol při použití aldehydového elektrofilu).[1][2] Reakci objevili Anthony B. Baylis a Melville E. D. Hillman, obdobnou reakci však popsal také K. Morita, proto bývá někdy nazývána Moritova–Baylisova–Hillmanova reakce nebo MBH reakce.[3]
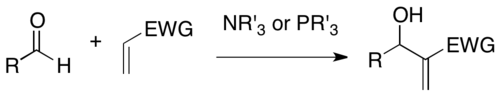
Jedním z nejčastějších katalyzátorů této reakce je DABCO, úspěšně byly použity také nukleofilní aminy jako jsou 4-dimethylaminopyridin (DMAP) a 1,8-diazabicyklo[5.4.0]undec-7-en (DBU) a též některé fosfiny.
Baylisova-Hillmanova reakce má několik výhod: 1) výchozí materiály lze připravit snadno. 2) Reakcí prochirálních elektrofilů se vytváří chirální centra, takže lze provést asymetrickou syntézu. 3) Produkty obvykle obsahují několik funkčních skupin blízko sebe, takže je možné provést řadu dalších přeměn. 4) Lze při ní použít nukleofilní organokatalyzátory bez těžkých kovů, a to za mírných podmínek.
Bylo vydáno několik prací zabývajících se touto reakcí.[4][5][6][7][8]
Mechanismus
Poprvé byl mechanismus Baylisovy-Hillmanovy reakce navržen v roce 1983.[9] Prvním krokem je 1,4-adice katalytického terciárního aminu na ativovaný alken za tvorby zwitteriontového azaenolátu. Ve druhém kroku se tento enolát naváže na aldehyd prostřednictvím aldolové reakce. Třetím krokem je vnitromolekulární přesun protonu, jehož produkt poté vytvoří konečný produkt a obnoví katalyzátor E2 nebo E1cb eliminací.[10] Závislost rychlosti reakce akrylonitrilu s acetaldehydem na jejich koncentracích a koncentraci DABCO je prvního řádu. Krokem určujícím rychlost je pravděpodobně aldolová adice, které se účastní všechny tři reaktanty. Kinetické izotopové efekty při použití α-deuterovaného akrylonitrilu nebyly pozorovány, což tento mechanismus podporuje.
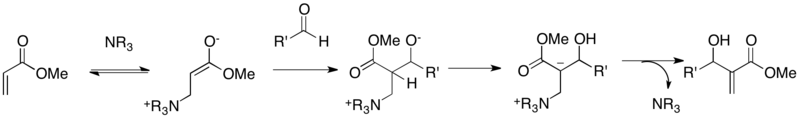
Tento první návrh však vykazoval několik nedostatků. Tvorba produktu reakci urychlovala (šlo o autokatalýzu), což tento mechanismus nedokázal vysvětlit; také se na jeho základě nedala očekávat tvorba dioxanonového vedlejšího produktu při reakcích arylaldehydů s akryláty.
McQuade et al. a Aggarwal et al. přepracovali mechanismus provedením kinetických a teoretických studií se zaměřením na přesun protonu.[11][12] McQuade et al. dospěli k závěru, že reakce methylakrylátu s p-nitrobenzaldehydem je vzhledem k aldehydu druhého řádu a vykazuje silný kinetický izotopový efekt (KIE) na pozici α v akrylátu. Nezávisle na rozpouštědle byl KIE větší než 2, což ukazovalo na to, že krokem určujícím rychlost je odštěpení protonu. Na základě těchto dat byl navržen nový mechanismus. První a druhý krok zůstaly stejné, ale po první aldolové adici následovala druhá adice aldehydu za vzniku poloacetalového alkoxidu. Následně proběhl přesun protonu přes šestičlenný meziprodukt a vytvořil se adukt A, jenž následně reagoval a vytvořil konečný produkt B nebo dioxanonový vedlejší produkt C.
Aggarwal se soustředil na autokatalytický efekt a zjistil, že katalytickými množstvími produktu nebo methanolu jej lze omezit. Navrhl tak mechanismus, na jehož začátku, nekatalyzovaném alkoholem, podobném McQuadeově variantě, kde po přeměně 20 % začne převažovat katalýza alkoholem. Alkohol R'OH zde ovlivňuje odštěpení protonu přes šestičlenný meziprodukt. Aggarwal a Harvey modelovali tyto mechanismy pomocí teorie funkcionálu hustoty a zjistili, že vypočtený energetický profil dobře odpovídá pozorovanému izotopovému kinetickému efektu i rychlosti reakce.[13] Celková entalpická bariéra byla o trochu nižší u dráhy katalyzované alkoholem, takže s růstem koncentrace alkoholu (produktu reakce) se zvyšuje podíl dráhy katalyzované alkoholem, a tedy dochází k autokatalýze.

I přes tyto studie existuje stále několik nedořešených otázek. McQuadeův předpoklad o vlivu meziproduktu A není potvrzen. Protože A se může vytvořit jednoduchou adicí B na aldehyd, tak ke vzniku A a C může docházet mimo tento mechanismus. McQuade předpokládá, že krok určující rychlost obsahuje dvě molekuly aldehydu, protože jde o reakci druhého řádu vzhledem k aldehydu, ovšem nevysvětluje Hillovo a Isaacovo pozorování prvního řádu u jimi použitých substrátů. Velká různorodost substrátů pro MBH reakce komplikuje zkoumání obecného mechanismu. Aggarwal také ukázal , že krokem určujícím rychlost je nejprve přesun protonu a později aldolová adice, protože primární kinetický izotopový efekt po 20% přeměně mizí,[12] ovšem následné výpočetní studie naznačily, že nejvyšší bariéru má i v závěrečné fázi reakce přesun protonu. Nesoulad mezi kinetickými a výsledky naznačuje, že stále existují součásti mechanismu MBH reakcí, které nejsou dobře prozkoumány.
Coelho a Eberlin et al. použili data z ESI-MS k získání experimentálních dat k podpoření dvojí povahy přesunu protonu při této reakci, čímž získali strukturální důkazy pro McQuadeovy a Aggarwalovy návrhy.[14]
U asymetrické MBH reakce
Aggarwalův model naznačil možnost provedení asymetrických MBH reakcí. Při reakci podle něj vznikají všechny čtyři diastereomery alkoxidového meziproduktu, ovšem pouze u jednoho je donor vodíkové vazby umístěn tak, aby umožňoval rychlý přenos protonu, ostatní diastereomery se mění zpět na výchozí látky. Tyto mechanistické studie obrátily pozornost na schopnost katalyzátoru (Brønstedovy kyseliny) poskytovat protony. Pokud má Brønstedova kyselina nebo Lewisova zásada vhodnou polohu vůči chirální molekule, tak Lewisova zásada bude reagovat se substrátem, zatímco kyselina umožní chirální přesun protonu. Brønstedova kyselina zůstává navázána na vzniklý enolát, ze kterého se poté vytvoří aldehyd, a zajišťuje tak účinný přenos protonu v kroku určujícím rychlost. Kokatalyzátor tvořený Brønstedovou kyselinou nemá vliv jen na přenos protonu, ale také vyvolává navázáním na zwitteriontový enolát konjugovanou adici a stabilizuje meziprodukty.
Rozsah
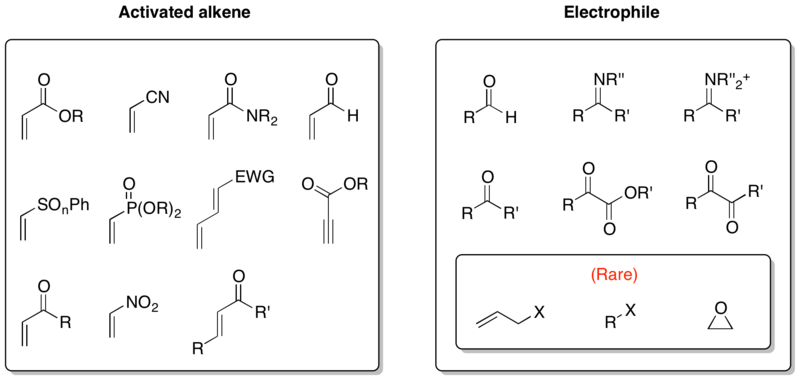
Protože jsou reaktanty při MBH reakcích obecné aktivované alkeny a elektrofily, tak lze použít značné spektrum látek. Obzvláště významným druhem je aza Baylisova–Hillmanova reakce, kde se jako elektrofily používají iminy. I když jsou elektrofily při MBH reakcích nejčastěji aldehydy, ketony nebo iminy, tak bylo popsáno i několik případů využívajících allylhalogenidy, alkylhalogenidy či epoxidy.[15][16][17]
Produkty Baylisových–Hillmanových reakcí a jejich deriváty lze použít k přípravě heterocyklů a jiných cyklických sloučenin.[18]
Při použití allenů místo jednoduchých alkenů probíhá reakce více na gama uhlících než v poloze alfa.[19]
Omezení
Vzhledem k velkému rozsahu použitelných substrátů je často obtížné najít vhodné reakční podmínky pro danou kombinaci substrátů, například β-substituované alkeny, vinylsulfony a vinylsulfoxidy mívají nízké reaktivity, samotná reakce je tak pomalá nebo vůbec neprobíhá. Potíže mohou způsobovat také vedlejší reakce funkčních skupin na substrátu. Akroleiny bývají náchylné k oligomerizacím a allenoáty snadno vstupují do cykloadicí. Obzvláště náročné je nalezení vhodných podmínek u alkylhalogenidových a epoxidových elektrofilů.
Pomalý průběh Baylisových-Hillmanových reakcí (s reakčními časy okolo dvou týdnů i delšími, i při použití 25 až 100 mol% katalyzátoru) u stericky stíněných alifatických aldehydů a benzaldehydů bohatých na elektrony, což omezuje její syntetickou využitelnost. Jako příklad lze uvést reakci t-butylakrylátu s benzaldehydem za katalýzy DABCO a nepřítomnosti rozpouštědla ppotřebovala k získání významného množství produktu 4 týdny. Při použití aprotických rozpouštědel je ještě pomalejší, ai když přídavek protických sloučenin (jako jsou alkoholy a karboxylové kyseliny) může reakci urychlit.[20]
Ketonyobvykle nejsou tak reaktivní, aby se mohly účastnit synteticky využitelných MBH reakcí. Přestože je míra aktivace u Baylisových-Hillmanových reakcí ketonů výrazně záporná a obyčejně tak reakce probíhá pomalu, tak je lze provést za vysokých tlaků (až 2 GPa).[4]
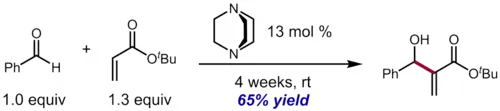
Vysoká reaktivita aktivovaných alkenů také může způsobovat potíže. MBH reakce arylvinylketonů nejsou jednoduché, protože reaktivní arylvinylketon se Michaelovou reakcí snadněji aduje na další molekulu arylvinylketonu a teprve poté vzniklý adukt reaguje s aldehydem na dvojitý MBH adukt.[21]
Obecné provedení asymetrické MBH reakce pro široké spektrum substrátů dosud není známo. Ohledně MBH reakcí je stále mnoho okolností, které by měly být vylepšeny, jako jsou například obecně použitelné katalytické systémy.
Varianty
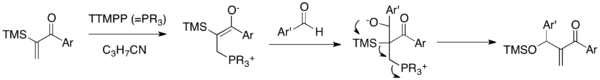
Sila-MBH reakce
Sila-MBH reakce používají α-silylované vinylarylketony a aldehydy za přítomnosti tris(2,4,6-trimethoxyfenyl)fosfinu (TTMPP)).[22] Zwitteriontový enolát, vytvořený adicí nukleofilního katalyzátoru na enon, se aduje na karbonylovou skupinu aldehydu za tvorby alkoxidu. Tento alkoxid následně projde 1,3-Brookovým přesmykem a eliminací se vytvoří siloxy-methylenenon a dojde k obnově katalyzátoru. Toto reakcí se dají připravit siloxy-methylenarylenones, sloučeniny, jež není možné vytvořit klasickými MBH reakcemi. Tato varianta se vypořádává s potížemi spojenými s arylvinylketony.
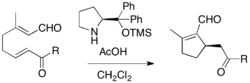
Rauhutova-Currierova reakce
V Rauhutových-Currierových reakcích, nazývaných také vinylogní MBH reakce, s aktivovaným alkenem reaguje Michaelův akceptor namísto aldehydu nebo iminu. Protože se zde často používají dva aktivované alkeny, tak bývají problémy se selektivitou. Vnitromolekulární Rauhutovy-Currierovy reakce mohou vykazovat lepší reaktivitu a selektivitu, například Rauhutovými-Currierovými cyklizacemi α,β-nenasycených aldehydů za přítomnosti kyseliny octové a derivátů prolinu se tvoří produkty se zvýšenou enantioselektivitou.[23]
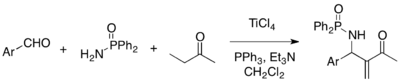
Tandemové a vícesložkové reakce
MBH reakce je možné pro vést jako třísložkové reakce aldehydů s aminy a aktivovanými alkeny za tvorby aza-MBH aduktů, příkladem mohou být reakce arylaldehydů, difenylfosfinamidů a methylvinylketonů za přítomnosti TiCl4, trifenylfosfinu a triethylaminu, jimiž se vytvářejí příslušné aza-MBH adukty.[24]
K elektrofilům lze přidat aktivované acetyleny a provést Michaelovu reakci. Jako Michaelův donor u třísložkových reakcí může sloužit mimo jiné trimethylsilyljodid; lze také uskutečnit tandemovou cyklizaci Michaelovým atakem MBH elektrofilu.[25]
Asymetrické Baylisovy-Hillmanovy reakce
Použití chirálních pomocníků
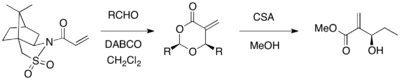
Oppolzerův sultam může být použit jako chirální pomocník pro asymetrické MBH reakce. Akrylát substituovaný Oppolzerovým sultamem reagoval s různými aldehydy (katalyzátorem byl DABCO), po odstranění chirálního pomocníka byly získány 1,3-dioxan-4-ony s 67-98% výtěžností a více než 99% enantiomerním přebytkem. Tyto cyklické produkty bylo možné přeměnit na pořadované produkty MBH pomocí kyseliny kamforsulfonové (CSA) a methanolu.[26]
Podobný hydrazidový pomocník může být použit na jinou, nepříliš odlišnou, MBH reakci katalyzovanou DABCO. Chirální akryloylhydrazid může diastereoselektivně reagovat s aldehydy.[27] Ze stejných reaktantů lze podle zvoleného rozpouštědla získat oba diastereomery (v DMSO vzniká jeden a ve směsi THF s vodou druhý), což naznačuje, že konformace přechodného stavu závisí na rozpouštědle.
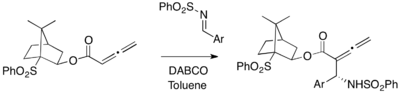
Chirální alleny a iminy lze zapojit do asymetrických aza-MBH reakcí.[28] Opticky aktivní 10-fenylsulfonylisobornyl-buta-2,3-dienoát reaguje s aryliminy, přičemž se diastereoselektivně vytváří α-allenylaminy (s výtěžností 37 až 57 %).
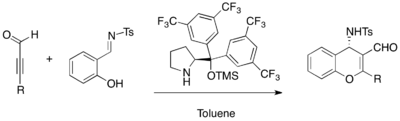
Katalýza chirálními Lewisovými zásadami
Enantioselektivní MBH reakce mohou být katalyzovány chirálními terciárními aminy. β-ICD, derivát cinchonového alkaloidu, patřící mezi chinidinové například katalyzuje reakce 1,1,1,3,3,3,-hexafluorisopropylakrylátu (jenž je aktivovaným alkenem) s mnoha různými aldehydy.[29] Fenolový kyslík β-ICD dává této sloučenině vlastnosti Brønstedovy kyseliny. β-ICD a podobné sloučeniny účinně katalyzují i reakce mnoha jiných substrátů.
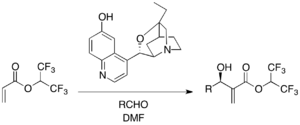
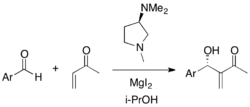
Cyklopentenon a řada dalších aromatických a alfatických aldehydů vstupuje do asymetrických reakcí za přítomnosti chirálního DMAP katalyzátoru v isopropylalkoholu; výtěžnosti se přitom pohybují mezi 54 a 96 % a enantiomerní přebytky od 53 do 98 %. K urychlení reakce je zde nutné použití Lewisovy kyseliny, konkrétně jodidu hořečnatého.[30] Jako možné katalyzátory zde byly rovněž zkoumány P-chirální fosfiny.[31]
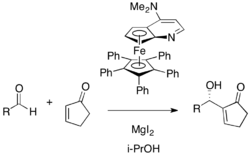
Lze použít i jednoduché diaminy. Methylvinylketon může reagovat se substituovanými benzaldehydy. Chirální pyrrolidinové katalyzátory vykazují vysokou účinnost u ortho- a para-substituovaných benzaldehydů chudých na elektrony (výtěžnosti 75-99 %, enantiomerní přebytky 8-73 %).[32]
Chirální fosfinové katalyzátory MBH reakcí mají na sobě často navázané skupiny fungující jako Brønstedovy kyseliny, například chirální fosfiny obsahující Lewisovy zásady, Brønstedovy kyseliny a kysele aktivované Brønstedovy zásady byly použity při asymetrických aza-MBH reakcích (výtěžnosti 86-96 %, enantiomerní přebytky 79-92 %). Skupiny fungující jako Brønstedovy kyseliny a zásady stabilizují zwitterionty stereoselektivním způsobem.[33]
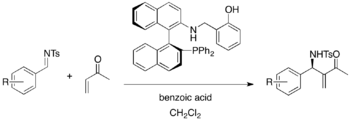
Chirální fosfiny odvozené od BINOLu mají také dobré katalytické vlastnosti a lze je použít pro aza-MBH reakce N-tosyliminů s aktivovanými alkeny, například methylvinylketonem a fenylakrylátem.[34]
Chirální fosfiny obsahující squaramidové skupiny mohou být použity na katalýzu vnitromolekulárních asymetrických Baylisových-Hillmanových reakcí, s ω-formylenony dávají cyklické enantiomerně obohacené produkty s výtěžnostmi od 64 do 98 % a enantiomerními přebytky mezi 88 a 93 %.[35]
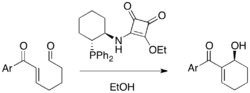
Chirální Lewisovy kyseliny
Chirální Lewisovy kyseliny mohou enantioselektivně aktivovat skupiny odtahující elektrony. Chirální oxazaborolidiniové kationty jsou dobré pro třísložkové reakce α,β-acetylenesterů s aldehydy a trimethylsilyljodidem, při výtěžnostech 50-99 % a enantiomerních přebytcích 62-94 %. Volbou enantiomeru katalyzátoru lze ovlivnit stereochemii produktu.[36]
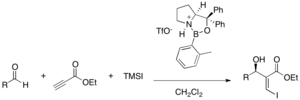
Použitelné mohou být i komplexy solí kovů s chirálními ligandy. La(OTf)3 a ligandy odvozené od kafru vytváří enantioselektivitu při MBH reakcích řady aldehydů a akrylátů katalyzovaných DABCO (výtěžnost 25-97 %, enantiomerní přebytek 6-95 %. K chelataci kovu se zde obvykle využívají multidentátní ligandy, jež aktivují jak zwitteriontový enolát, tak také aldehyd.[37]
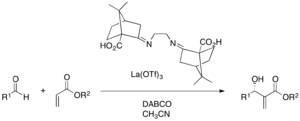
Systémy složené z La(O-iPr)3 a BINOLu, ve spojení s DABCO jako katalyzátorem, mohou být použity na mnohé asymetrické aza-MBH reakce řady různých N-difenylfosfinoyliminů s methylakrylátem. Aryl-, heteroaryl- a alkenyliminy jsou vhodné díky vysokým výtěžnostem i dobrým hodnotám enantioselektivity.[38]
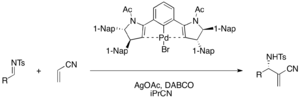
Chirální palladnaté komplexy lze použít jako Lewisovy kyseliny při enantioselektivních aza-MBH reakcích akrylonitrilu s tosyliminy, při kterých vznikají funkcionalizované α-methylen-β-aminonitrily s výtěžnostmi mezi 75 a 98 % a enantiomerními přebytky mezi 76 a 98 %. K aktivaci prekatalyzátoru tvořeného bromidem palladnatým se používá octan stříbrný.[39]
Chirální Brønstedovy kyselina jako kokatalyzátory
V souvislosti s asymetrickými Baylisovými-Hillmanovými reakcemi byla také zkoumána katalýza chirálními thiomočovinami. Chirální thiomočoviny a bis(thiomočoviny) mohou být použity jako kotalyzátory MBH a aza-MBH reakcí katalyzovaných pomocí DABCO.[40][41] Jacobsenovým katalyzátorem lze katalyzovat například enantioselektivní aza-Baylisovy-Hillmanovy reakce.
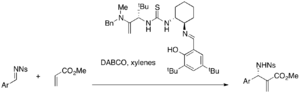
Zatímco jednoduché thiomočoviny vyžadují současnou přítomnost nukleofilního katalyzátoru, tak bifunkční katalyzátory, jako jsou fosfin-thiomočoviny , lze použít samotné; například akryláty reagují za jejich přítomnosti s aromatickými aldehydy a vytváří se přitom produkty enantioselektivních MBH reakcí (při výtěžnostech od 32 do 96 % a enantiomerních ořebytcích od 9 do 77 %.[42]
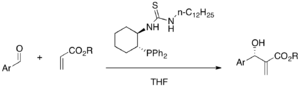
Kokatalyzátory MBH reakcí mohou být odvozené od prolinu. Imidazolový nukleofil a prolin vedou reakci skrze iminiové meziprodukty.[43] Za použití (S)-prolinu a DABCO reagují α-amidosulfony s α,β-nenaycenými aldehydy za vysoce enantioselektivních aza-MBH reakcí.[44]
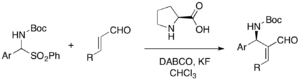
Využití
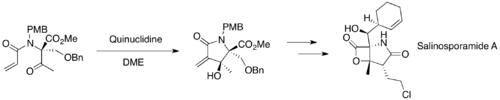
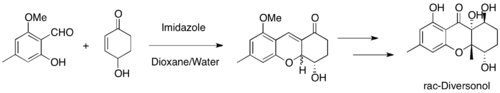
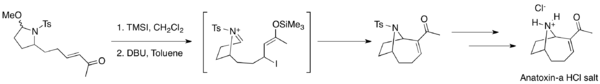
MBH reakce jsou v organické syntéze časté, například se používají k vytváření cyklických meziproduktů při syntézách salinosporamidu A, diversonolu a anatoxinu A.[45][46][47]
Reference
V tomto článku byl použit překlad textu z článku Baylis–Hillman reaction na anglické Wikipedii.
- Baylis, A. B.; Hillman, M. E. D. German Patent 2155113, 1972.
- Ciganek, E. Organic Reactions 1997, 51, 201. DOI:10.1002/0471264180.or051.02
- K. Morita, Z. Suzuki and H. Hirose, Bulletin of the Chemical Society of Japan,1968, 41, 2815
- Recent Advances in the Baylis−Hillman Reaction and Applications Deevi Basavaiah, Anumolu Jaganmohan Rao, and Tummanapalli Satyanarayana Chemical Reviews, 2003, 103 (3), pp 811–892 2003 (Article) DOI:10.1021/cr010043d
- Masson, G., Housseman, C. and Zhu, J. (2007), The Enantioselective Morita–Baylis–Hillman Reaction and Its Aza Counterpart. Angewandte Chemie International Edition, 46: 4614–4628. DOI:10.1002/anie.200604366
- aza-Baylis−Hillman Reaction Valerie Declerck, Jean Martinez and Frederic Lamaty Chemical Reviews, 2009, 109 (1), pp 1–48, 2009 (Review) DOI:10.1021/cr068057c
- Recent Contributions from the Baylis−Hillman Reaction to Organic Chemistry Deevi Basavaiah, Bhavanam Sekhara Reddy and Satpal Singh Badsara Chemical Reviews 2010 110 (9), 5447-5674 DOI:10.1021/cr900291g
- The Baylis–Hillman reaction: a novel concept for creativity in chemistry Deevi Basavaiah and Gorre Veeraraghavaiah Chemical Society Reviews, 2012, Advance Article DOI:10.1039/C1CS15174F
- Angew. Chem. Int. Ed. Engl. 1983, 22, 795.
- J. Phys. Org. Chem. 1990, 3, 285
- Organic Letters, 2005, 7, 1, 147-150.
- Angew. Chem. Int. Ed. 2005, 44, 1706-1708
- Journal of the American Chemical Society 2007, 129, 15513
- The Journal of Organic Chemistry, 2009, 74(8), 3031-3037
- Tetrahedron Lett. 2001, 42, 85.
- Org. Lett. 2010, 12, 2418.
- Chem. Commun. 2006, 2977.
- Tetrahedron, 2008, 64(20), 4511-4574.
- J. Am. Chem. Soc. 2009, 131, 4196.
- Yves Fort; Marie Christine Berthe; Paul Caubere. The 'Baylis - Hillman Reaction' mechanism and applications revisited. Tetrahedron. 1992, s. 6371–6384. DOI 10.1016/s0040-4020(01)88227-2.
- Angew. Chem. Int. Ed. 2012, 51, 10337.
- Organic Letters, 2009, 11, 1, 253-255.
- Org. Lett. 2009, 11, 4116.
- Tetrahedron Lett., 2002, 43, 9171.
- Chem. Eur. J. 2010, 16, 9453
- Journal of the American Chemical Society 1997, 119, 4317-4318
- Organic Letters 2000, 2, 6, 729-731
- European Journal of Organic Chemistry 2010, 3249-3256
- Journal of the American Chemical Society 1999, 121, 10219-10220
- Chemical Communications 2010, 46, 2644-2646
- Y. Xiao; Z. Sun; H. Guo; O. Kwon. Chiral Phosphines in Nucleophilic Organocatalysis. Beilstein Journal of Organic Chemistry. 2014, s. 2089–2121. DOI 10.3762/bjoc.10.218. PMID 25246969.
- J. Tetrahedron: Asymmetry, 2010, 1511.
- Advanced Synthesis & Catalysis 2009, 351, 331
- Chemical Communications 2003, 1310
- Chemical Communications 2011, 47, 1012
- Angewandte Chemie International Edition 2009, 48, 4398
- The Journal of Organic Chemistry 2003, 68, 915-919
- Journal of the American Chemical Society 2010, 132, 11988
- Angewandte Chemie International Edition 2012, 51, 10337-10341
- Advanced Synthesis & Catalysis 2005, 347, 1701-1708
- Tetrahedron Letters 2011, 52, 6234
- Tetrahedron 2009, 65, 8185
- Chemistry—A European Journal 2009, 15, 1734
- Advanced Synthesis & Catalysis 2011, 353, 1096
- Journal of the American Chemical Society 2004, 126, 6230-6231.
- Angewandte Chemie International Edition 2006, 45, 307–309.
- Chemical Communications 2008, 3432