Nukleofilní acylová substituce
Nukleofilní acylová substituce je pojem označující skupinu substitučních reakcí, při kterých reagují nukleofily se sloučeninami obsahujícími acylové skupiny. Nukleofil, jako například alkohol, amin nebo enolát, vyvolává odštěpení acylové skupiny (například z acylhalogenidu, amidu nebo esteru). Produktem je karbonylová sloučenina, ve které nukleofil nahradil odcházející skupinu původní acylové sloučeniny. Acylové sloučeniny reagují s mnoha různými nukleofily a protože konkrétní produkt může být závislý na druhu použité acylové aloučeniny a nukleofilu, tak lze tímto způsobem připravit velké množství sloučenin.
Mechanismus
Karbonylové sloučeniny reagují s nukleofily adičním mechanismem: nukleofil atakuje karbonylový uhlík za vzniku čtyřstěnného meziproduktu. Tento krok je možné urychlit okyselením reakční směsi, které vede ke zvýšení elektrofility karbonylové skupiny, nebo přidáním zásady, čímž se nukleofil stane více aniontovým a tím i reaktivnějším. V závislosti na pH v reakčním prostředí může být meziproduktem alkohol nebo alkoxid.
V meziproduktu se nachází substituent, který je navázán na centrální uhlík a může se stát odcházející skupinou. Meziprodukt se po svém vzniku přemění na konečný produkt za obnovení karbonylové skupiny a odštěpení odcházející skupiny při eliminační reakci. Výsledkem tohoto procesu je nahrazení odcházející skupiny nukleofilem způsobem odpovídajícím stavu, kdy by meziprodukt neobsahoval karbonylovou skupinu. Oba kroky jsou vratné a mezi produkty bude vždy převažovat lepší nukleofil a k tomu, aby měla tato reakce význam, tak musí být odcházející skupina špatným nukleofilem.
V kyselém prostředí
V kyselém prostředí je karbonylová skupina acylové sloučeniny 1 protonována, což ji aktivuje vůči nukleofilním atakům. Ve druhém kroku je protonovaný karbonyl 2 atakován nukleofilem (H−Z) za vzniku tetraedrického meziproduktu 3. Přenos protonu z nukleofilu (Z) na odcházející skupinu (X) vede ke vzniku meziproduktu 4, který odštěpuje protonovanou odcházející skupinu (H−X) a vytváří tak protonovanou karbonylovou sloučeninu 5. Z té deprotonací vzniká konečný produkt 6. Nukleofilní acylové substituce se považují za kysele katalyzované reakce, protože při posledním kroku dochází k odštěpení protonu. Nukleofil se v kyselém prostředí obvykle vyskytuje v protonované formě (H-Z namísto Z−).

V zásaditém prostředí
V zásaditém prostředí nukleofil atakuje karbonyl acylové sloučeniny 1 za vzniku alkoxidového meziproduktu 2. Z tohoto meziproduktu se odštěpuje odcházející skupina (X) a tvoří se konečný produkt 3. I když může být nukleofilní acylová substituce katalyzovaná zásadou, tak reakce nebude probíhat, pokud je odcházející skupina slabší zásadou než nukleofil (musí tedy mít vyšší pKa). Na rozdíl od kysele katalyzované reakce se zde nukleofil i odcházející skupina vyskytují v aniontových formách.

Průběh reakce tímto způsobem podporují výsledky experimentů s izotopovým značkováním. Při reakci ethyl-propanoátu s ethoxyskupinami značkovanými kyslíkem-18 byla kyselina propionová zbavena tohoto izotopu, který zůstal pouze v molekulách ethanolu.[1]
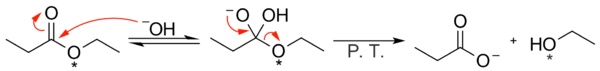
Reaktivita různých substrátů
Je celkem pět hlavních druhů acylových sloučenin. S nukleofily nejlépe reagují acylhalogenidy, následují anhydridy, estery a amidy. Karboxylátové ionty nereagují, protože nemají odpovídající odcházející skupinu. Reaktivita acylchloridů a amidů se liší o 13 řádů.[2]
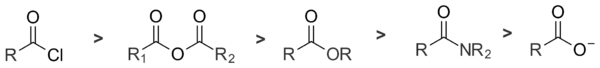
Na reaktivitu acylových sloučenin má největší vliv schopnost vytvořit odcházející skupinu. Látky, které jsou slabšími zásadami, a mají tedy silnější konjugované kyseliny, jsou lepšími odcházejícími skupinami; například chloridový anion reaguje lépe než octanový anion. S rostoucí zásaditostí se reaktivita acylových sloučenin v nukleofilních substitucích snižuje:[3]
| Sloučenina | Strukturní vzorec | Odcházející skupina | pKa konjugované kyseliny |
|---|---|---|---|
| Acetylchlorid | 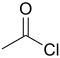 |
−7 | |
| Acetanhydrid | 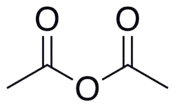 |
 |
4,76 |
| Ethylacetát | 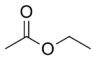 |
15,9 | |
| Acetamid | 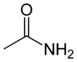 |
38 | |
| Octanový anion | |
není | není |
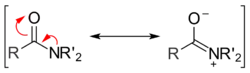
Amidy karboxylových kyselin mají dvě hlavní rezonanční struktury. Energetická bariéra rotace kolem amidové vazby bývá 75–85 kJ/mol, což je mnohem více než u běžných jednoduchých vazeb; například u vazby C-C v molekule ethanu je to kolem 12 kJ/mol.[2] Poté, co nukleofil atakuje substrát a vznikne tetraedrický meziprodukt, tak energetická výhodnost rezonančních struktur zmizí; tímto lze vysvětlit velmi nízkou reaktivitu amidů.[3]
U esterů je rezonanční stabilizace slabší než u amidů, takže tvorba tetraedrického meziproduktu a následná ztráta rezonančních struktur je energeticky nevýhodná. Anhydridy mají rezonanční stabilizaci ještě slabší, protože je rozdělena mezi dvě karbonylové skupiny, a jsou tak reaktivnější než estery. U acylhalogenidů je rezonance nevýrazná, a tak je energetická bariéra tvorby tetraedrického meziproduktu velmi nízká, což vysvětluje, že jsou ze všech acylových derivátů nejreaktivnější.[3]
Reakce acylových sloučenin
Při mnoha nukleofilních acylových substitucích dochází k přeměně jedné acylové sloučeniny v jinou. Aby měly takovéto reakce praktický význam, tak musí z výrazně reaktivní sloučeniny vznikat málo reaktivní, například acylchloridy lze snadno přeměnit na estery, ovšem opačná přeměna je neproveditelná. Produkt by měl být stabilnější než reaktant.
Mohou také probíhat nukleofilní acylové substituce, při kterých nedochází k přeměně jednoho acylového derivátu v jiný. Amidy a karboxylové kyseliny mohou například reagovat s Grignardovými činidly za vzniku ketonů.
Acylhalogenidy
Acylhalogenidy jsou nejreaktivnějšími acylovými sloučeninami a mohou být snadno přeměněny na jiné. S karboxylovými kyselinami vytvářejí anhydridy. Nejprve karboxylová kyselina atakuje acylhalogenid 1 za vzniku čtyřstěnného meziproduktu 2. Z toho se oddělí chloridový anion a vytvoří se oxoniová sloučenina 3. Z ní se deprotonací utvoří anhydrid 4 a dojde k odštěpení halogenvodíku.

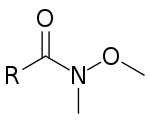
Na rozdíl od ostatních uhlíkatých nukleofilů mohou lithné dialkylměďnatany (také nazývané Gilmanova činidla) reaguvat s acylhalogenidy za vzniku ketonů v jediné reakci. Tato reakce nepatří mezi nukleofilní acylové substituce, ale probíhá radikálovým mechanismem.[1] K přeměně acylhalogenidů na ketony Weinrebovu syntézu ketonů. Při této reakci se nejprve acylhalogenid přemění na N–methoxy–N–methylamid, známý také jako Weinrebův amid. Po adici uhlíkatého nukleofilu, například organolithné sloučeniny nebo Grignardova činidla, dojde k chelataci kovu karbonylovým a N–methoxy kyslíkem, čímž se zabrání dalším nukleofilním adicím.
Thioestery
Thioestery reagují podobně jako alkylhalogenidy, pouze poněkud pomaleji.
Anhydridy
Anhydridy karboxylových kyselin reagují podobně jako acylhalogenidy. Nelze je přímo přeměnit na acylhalogenidy, ale je možné z nich připravit ostatní acylové deriváty. Mohou se účastnit Schottenových–Baumannových reakcí, kdy se používají k odstraňování esterů a amidů z alkoholů a aminů a rovněž reagují s vodou na příslušné karboxylové kyseliny. Podobně jako acylhalogenidy reagují s karbonylovými nukloefily a lze je použít při Friedelových–Craftsových reakcích a přípravě Weinrebova ketonu. Na rozdíl od acylhalogenidů však nereagují s Gilmanovými činidly.[1]
Reaktivitu anhydridů je možné zvýšit přidáním katalytického množství N,N-dimethylaminopyridinu (DMAP) nebo pyridinu.

Na začátku anhydrid 1 reaguje s DMAP 2 na čtyřstěnný meziprodukt, ten se následně odštěpením karboxylátového iontu přemění na amid 3. Tento amid je lépe aktivován vůči nukleofilním atakům, protože dimethylaminopyridin je lepší odcházející skupinou než karboxylát. 3 poté reaguje s nukleofilem za vzniku dalšího čtyřstěnného meziproduktu, který se přemění na konečný produkt 4; dojde přitom k eliminaci pyridinové skupiny a obnovení její aromaticity.
Estery
Estery jsou méně reaktivní než acylhalogenidy a anhydridy. Reagují s amoniakem, primárními i sekundárními aminy za vzniku amidů, tento způsob přípravy se však nepoužívá často, protože reakce amoniaku a aminů s acylhalogenidy mívají vyšší výtěžnost. Estery lze přeměňovat na další estery transesterifikací, tedy reakcí esteru s alkoholem; ta může být kysele i zásaditě katalyzovaná. Odcházející skupinou je zde ovšem alkohol a přímá i zpětná reakce tak budou probíhat podobně rychle. Pomocí výrazného přebytku alkoholu použitého jako reaktant nebo odstraňováním vznikajícího alkoholu (například destilací) se dosáhne převahy přímé reakce.
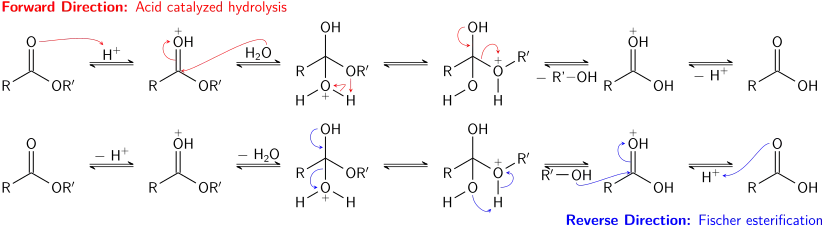
Při zásadité hydrolýze esterů dochází k úplné spotřebě zásady a vzniku jednoho ekvivalentu alkoholu a jednoho ekvivalentu karboxylátové soli. Tímto způsobem se z esterů mastných kyselin vyrábějí mýdla.
Estery mohou podstupovat řadu různých reakcí s uhlíkatými nukleofily. Podobně jako acylhalogenidy a anhydridy reagují s Grignardovými činidly za vzniku terciárních alkoholů. Také snadno reagují s enoláty. Při Claisenově kondenzaci enolát jednoho esteru atakuje karbonylovou skupinu druhého esteru 2, čímž se tvoří meziprodukt 3, který odštěpí alkoxyskupinu a vytvoří β-ketoester 4.

Lze provést Claisenovy kondenzace, při kterých je jako nukleofil použit jiný ester, než od kterého je odvozen enolát. Vnitromolekulární obdoba Claisenovy kondenzac se nazývá Dieckmann kondenzace a používá se k přípravě cyklických sloučenin. Estery mohou rovněž kondenzovat s enoláty ketonů a aldehydů na β-dikarbonylové sloučeniny.[4] Zvláštním případem takovéto reakce je Bakerův–Venkataramanův přesmyk, kdy u aromatických ortho-acyloxyketonů proběhne vnitromolekulární nukleofilní acylová substituce a poté přesmyk, přičemž se vytvoří aromatický β-diketon.[5] Dalším případem přesmyku po vnitromolekulární nukleofilní acylové substituci je Chanův přesmyk.
Amidy
Vzhledem ke své nízké reaktivitě se amidy karboxylových kyselin účastní mnohem menšího počtu reakcí než ostatní acylové sloučeniny. Ve vodě jsou astabilní a jsou asi 100× odolnější vůči hydrolýze než estery.[2] Mohou být ovšem hydrolyzovány na karboxylové kyseliny za přítomnosti zásad nebo kyselin. Stabilita peptidové vazby má svůj biologický význam, protože bílkoviny se skládají z aminokyselin spojených právě tímto druhem vazeb. Jsou dostatečně stabilní na to, aby udržely strukturu bílkovin i ve vodném prostředí, ale dostatečně slabé na to, aby mohly být v případě potřeby narušeny.
Reakce primárních a sekundárních amidů s uhlíkatými nukleofily neprobíhají dobře. Grignardova činidla a organolithné sloučeniny se chovají více jako zásady než jako nukleofily a pouze amidovou skupinu deprotonují. Terciární amidy oproti tomu reagují za vzniku ketonů; amidový anion (NR2−) je velmi silnou zásadou a tedy není dobrou odcházející skupinou. N,N'-dimethylformamid lze použít k zavedení formylové skupiny do sloučenin reakcí s uhlíkatými nukleofily.[6]

Fenyllithium 1 atakuje karbonylovou skupinu na DMF 2 za vzniku čtyřstěnného meziproduktu 3. Protože je dimethylamidový anion špatnou odcházející skupinou, tak k další nukleofilní adici nedochází. V kyselém prostředí je alkoxid protonován na 4 a amin na 5. Eliminací dimethylaminu a ztrátou protonu vznikne benzaldehyd 6.
Karboxylové skupiny
Karboxylové kyseliny nejsou dobrými reaktanty pro nukleofilní substituce, protože je nelze přímo přeměnit na ostatní acylové sloučeniny. Přeměna na amid je možná, ovšem nelze ji provést v jediném kroku. Spíše než jako nukleofily budou s nimi aminy reagovat jako zásady a tvořit příslušné soli. Zahřívání těchto solí nad 100 °C vede k odštěpení vody a vzniku amidu. Tímto způsobem se získávají amidy v průmyslu i v laboratořích. Za přítomnosti silné kyseliny mohou karboxylové kyseliny podstoupit kondenzační reakce, při kterých se tvoří příslušné anhydridy karboxylových kyselin, které se však mohou hydrolyzovat na původní kyseliny.
Za přítomnosti silných kyselin reagují karboxylové kyseliny s alkoholy nebo fenoly na estery; k přípravě methylesterů je rovněž možné použít diazomethan.
S chloridem thionylu reagují karboxylové kyseliny za tvorby odpovídajících acylchloridů. Nejprve karboxylová kyselina 1 atakuje thionylchlorid za odštěpení chloridového iontu. Vzniklý oxoniový ion 2 je aktivován vzhledem k nukleofilním atakům a je dobrou odcházející skupinou, na rozdíl od běžné karboxylové kyseliny. V následujícím kroku 2 reaguje s chloridovým iontem a vytváří čtyřstěnný meziprodukt 3. Ten poté odštěpí oxid siřičitý a chloridový ion za vzniku protonovaného acylchloridu 4. Chloridový ion deprotonuje acylchlorid na konečný produkt 5, přičemž se přemění na chlorovodík.

Acylchloridy se také dají připravit reakcí karboxylových kyselin s chloridem fosforitým (PCl3) nebo chloridem fosforečným (PCl5), mechanismus je podobný. Jeden ekvivalent PCl3 může zreagovat se třemi ekvivalenty kyseliny, sám se přitom změní na kyselinu fosforitou. PCl5 reaguje s karboxylovými kyselinami v molárním poměru 1:1 a vytváří oxychlorid fosforečný (POCl3) a chlorovodík (HCl) jako vedlejší produkty.
Karboxylové kyseliny reagují s Grignarsovými a organolithnými činidly, přičemž vznikají ketony. První ekvivalent nukleofilu funguje jako zásada a deprotonuje kyselinu, zatímco druhý ekvivalent atakuje karboxylovou skupinu za tvorby alkoxidového dianiontu, který je následně protonován a vytváří hydrát ketonu. Většina hydrátů ketonů je značně nestabilních, a tak v rovnovážném stavu výrazně převažuje keton; například rovnovážná konstanta tvorby hydrátu acetonu z acetonu je 0,002.
Odkazy
Externí odkazy
 Obrázky, zvuky či videa k tématu Nukleofilní acylová substituce na Wikimedia Commons
Obrázky, zvuky či videa k tématu Nukleofilní acylová substituce na Wikimedia Commons - Reakce acetanhydridu s acetonem v Organic Syntheses Coll. Vol. 3, p. 16; Vol. 20, p. 6 Article
Reference
V tomto článku byl použit překlad textu z článku Nucleophilic acyl substitution na anglické Wikipedii.
- John McMurry. Organic Chemistry. Pacific Grove, CA: Brooks/Cole Publishing Company, 1996. Dostupné online. ISBN 0534238327. S. 820–821.
- Francis A. Carey. Organic Chemistry. New York: McGraw-Hill, 2006. Dostupné online. ISBN 0072828374. S. 866–868.
- Wade 2010, pp. 998–999.
- Carey 2006, pp. 919–924.
- Kürti and Czakó 2005, p. 30.
- Comprehensive Organic Functional Group Transformations. Příprava vydání Alan R. Katritzky, Otto Meth-Cohn, Charles Rees. Oxford: Pergamon Press, 1995. [=https://archive.org/details/comprehensiveorg0000unse/page/90 Dostupné online]. ISBN 0080423248.