Kumadovo párování
Kumadovo párování je druh křížové párovací reakce používaný na tvorbu nových vazeb uhlík–uhlík reakcí Grignardova činidla s organohalogenidem. Jako katalyzátory se používají přechodné kovy, většinou nikl nebo palladium, přičemž spolu mohou reagovat alkylové, arylové nebo vinylové skupiny. Reakci popsali v roce 1972 skupiny výzkumníků, které vedli Robert Corriu a Makoto Kumada.[1][2]

Tato reakce je jedním z prvních vyvinutých katalytických křížových párování. I přes rozvoj dalších reakcí (Suzukiovy, Sonogaširovy, Stilleovy, Hijamovy, Negišiovy) je Kumadova reakce stále zapojována do řady syntetických postupů, jako je například výroba aliskirenu, léku proti zvýšenému krevnímu tlaku, a polythiofenů, používaných v organické elektronice.
Historie
První výzkum katalytického párování Grignardových činidel s organohalogenidy provedli Morris S. Kharasch a E. K. Fields v roce 1941; použili při tom katalyzátory obsahující kobalt.[3]
V roce 1971 Masuhiko Tamura a Jay K. Kochi vydali několik článků ukazujících na použitelnost katalyzátorů založených na stříbru,[4] mědi[5] a železu.[6]
Tyto reakce měly ovšem nízké výtěžnosti, protože se pokaždé vytvářelo homopárováním významné množství vedlejších produktů, pokud byly k párování použity molekuly stejné sloučeniny.
V roce 1972 bylo popsáno využití niklových katalyzátorů. Po zavedení palladiových katalyzátorů v roce 1975 byla využitelnost reakce dále rozšířena.[7]
Poté se objevila řada dalších párovacích reakcí a v roce 2010 získali Eiči Negiši, Akira Suzuki a Richard F. Heck Nobelovu cenu za chemii.
Mechanismus
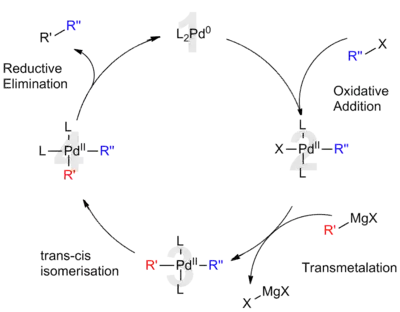
Při katalýze palladiem
V souladu s obecně příjímaným mechanismem se předpokládá, že u Kumadových párování katalyzovaných palladiem je význam palladia obdobný jako u jiných křížových párování. Navržený katalytický cyklus obsahuje palladium v oxidačních číslech 0 i II. Na začátku se na elektrony bohatý Pd0 katalyzátor (1) naváže na vazbu R–X v molekule organohalogenidu. Touto oxidační adicí se utvoří organopalladnatý komplex (2). Poté proběhne transmetalační reakce s Grignardovým činidlem za vzniku heteroorganokovového komplexu (3). Před následujícím krokem musí proběhnout izomerizace, aby byly sousední ligandy vzájemně v poloze cis. Nakonec se redukční eliminací meziproduktu (4) vytvoří vazba uhlík–uhlík, uvolní výsledný produkt a obnoví Pd0 katalyzátor (1).[8] U palladiových katalyzátorů rychlost reakce určuje oxidační adice, která probíhá pomaleji než u katalýzy niklem.[8]
Při katalýze niklem
Mechanismus párování katalyzovaných niklem není zcela znám. Předpokládá cse, že za různých reakčních podmínek a při použití odlišných ligandů probíhá rozdílně.[9] Obvykle se přesto popisuje jako podobný mechanismu u palladia. Za určitých podmínek ovšem takový mechanismus nedokáže vysvětlit všechny pozorované jevy. Při zkoumání reakcí tridentátních terpyridinových ligandů byly zjištěny mezipodukty vykazující katalytické cykly typu NiII-NiI-NiIII,[10] což naznačuje složitější průběh. Při přidání butadienu se nejspíše tvořil meziprodukt obsahující NiIV.[11]
Možnosti
Organické halogenidy a pseudohalogenidy
Kumadovo párování bylo úspěšně použito na velký počet aryl- i vinylhalogenidů. Místo halogenidů lze také použít pseudohalogenidy, například tosyláty[12] a trifláty,[13] a to za mnoha různých podmínek.
I přes úspěšná použití aryl- a vinylhalogenidů se alkylhalogenidy tolik neosvědčily. Protože nemají π-elektrony, tak alkylhalogenbidy vyžadují odlišné mechanismy oxidační adice než arylové a vinylové skupiny, tyto procesy nejsou dobře popsané.[9] Kvůli přítomnosti β-vodíků jsou také alkylhalogenidy náchylné k beta-hydridovým eliminacím.[14]
Tyto potíže byly překonány přidáním aktivujících skupin, například karbonylů v podobě α-bromketonů, které reakci usnadňují. Kumadova párování byla také provedena u neaktivovaných alkylů, často přidáním dalšího katalyzátoru nebo reaktantu; například za přítomnosti buta-1,3-dienů bylo možné uskutečnit niklem katalyzované alkyl–alkylové párovací reakce, které by jinak neprobíhaly.[15]
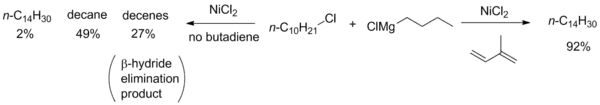
Předpokládá se, že mechanismus těchto reakcí zahrnuje tvorbu oktadienylového komplexu niklu. Tento katalyzátor by měl vstupovat do transmetalací s Grignardovými činidly a poté se předpokládá redukční eliminace halogenidu, čímž se omezuje nebezpečí β-hydridové eliminace. U párování arylů a vinylů byly zaznamenány produkty obsahující NiIV, který není v souladu s mechanismy navrženými pro párování arylových a vinylových halogenidů.[11]
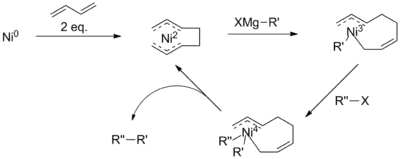
Grignardova činidla
Byla popsána párování aryl- a vinylGrignardových činidel.[2] Rovněž je možné bez větších obtíží použít alkylová Grignardova činidla, protože u nich neprobíhají β-hydridové eliminace. Přestože je za běžných podmínek možné s Grignardovými činidly použít jen omezené spektrum funkčních skupin, tak byly provedeny i reakce, do kterých lze zapojit větší počet různých substituentů, za nízkých teplot byly provedeny i reakce substrátů s vysoce funkcionalizovanými arylovými skupinami.[16]
Katalyzátory
Kumadovy párovací reakce může katalyzovat velký počet nikelnatých i palladnatých katalyzátorů. Struktury prekurzorů lze obecně popsat vzorcem ML2X2, kde L je fosfinový ligand.[17] Jako L2 se často používají bidentátní difosfiny, jako jsou dppe a dppp.
Alois Fürstner se svými spolupracovníky provedl reakce katalyzované komplexy železa, výtěžnosti. Pravděpodobnými aktivními katalyzátory jsou zde „anorganická Grignardova činidla“ se strukturou typu Fe(MgX)2.[18]
Reakční podmínky
Tyto reakce nejčastěji probíhají v tetrahydrofuranu nebo diethyletheru. Etherové roztoky jsou výhodné, protože jde o obvyklé prostředí, ve kterém se připravují Grignardova činidla.[2] Vzhledem k vysoké reaktivitě Grignardových činidel je spektrum funkčních skupin použitelných v Kumadových reakcích omezené, což může komplikovat větší syntézy. Grignardova činidla snadno podléhají protonolýzám za přítomnosti i mírně kyselých skupin, jako jsou například alkoholy. Také se mohou adovat na karbonylové i jiné oxidující skupiny.
Podobně jako u řady dalších párovacích reakcí jsou palladiové katalyzátory citlivé na přítomnost vzduchu, reakce se tak provádějí v inertních atmosférách tvořených argonem nebo dusíkem.
Selektivita
Stereoselektivita
Jak cis-, tak i trans-alkenylhalogenidy mohou vstupovat do reakcí s Grignardovými činidly zachovávajících výchozí geometrické konfigurace. Toto není závislé na ostatních faktorech, jako jsou ligandy u katalyzátoru a vinylové substituenty.[17]
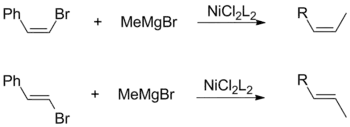
Kumadova párování využívající vinylová Grignardova činidla nemají žádnou stereospecificitu a vytváří směsi cis- and trans-alkenů. Míra izomerizace závisí na mnoha různých faktorech, jako jsou vlastnosti reaktantů a druh halogenidové skupiny. Tuto ztrátu stereospecifity způsobují vedlejší reakce mezi dvěma ekvivalenty allylových Grignardových činidel.[17]

Enantioselektivita
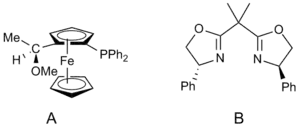
Asymetrické Kumadaovy párovací reakce lze uskutečnit při použití chirálních ligandů. U rovinně chirálních ferrocenových ligandů byly dosaženy enantiomerní přebytky i nad 95 %, pokud reagovaly aryly.[19]
Gregory Fu předvedl enantiokonvergentní párování α-bromketonů s katalyzátory obsahujícími bisoxazolinové ligandy, kde chirální katalyzátor přeměňuje výchozí racemickou směs na jediný enantiomer s enantiomerním přebytkem 95 %.[20]
Tato reakce je významná tím, že se při ní používá jinak většinou neproveditelné párování alkylhalogenidů.
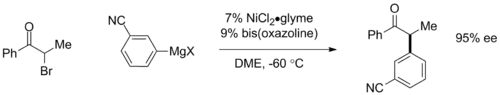
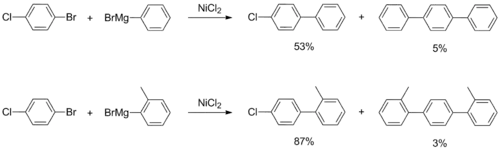
Použití
Syntéza aliskirenu
Kumadovy párovací reakce se používají i v průmyslovém měřítku, například při výrobě léčiv. Slouží mimo jiné k tvorbě uhlíkové kostry aliskirenu, léku proti vysokému krevnímu tlaku.[22]
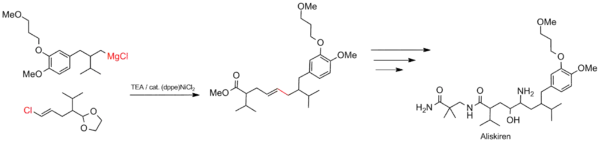
Syntéza polythiofenů
Kumadovo párování je také využitelné při tvorbě konjugovaných polymerů, jako jsou například polyalkylthiofeny (PAT), které mohou být použity v organických fotovoltaických článcích a světelných diodách.[23]
V roce 1992 byla uskutečněna první syntéza regiopravidelných polyalkylthiofenů, s využitím níže znázorněné Kumadovy reakce, která vyžadovala teploty pod 0 °C.[24]
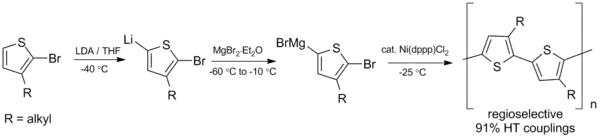
Tato syntéza byla následně vylepšena, za dosažení vyšší výtěžnosti a proveditelnosti při pokojové teplotě.
Odkazy
Externí odkazy
Související články
Reference
V tomto článku byl použit překlad textu z článku Kumada coupling na anglické Wikipedii.
- R. J. P. Corriu; J. P. Masse. Activation of Grignard reagents by transition-metal complexes. A new and simple synthesis of trans-stilbenes and polyphenyl. Journal of the Chemical Society, Chemical Communications. 1972, s. 144a. DOI 10.1039/C3972000144A.
- Kohei Tamao; Koji Sumitani; Makoto Kumada. Selective carbon–carbon bond formation by cross-coupling of Grignard reagents with organic halides. Catalysis by nickel-phosphine complexes. Journal of the American Chemical Society. 1972-06-01, s. 4374–4376. DOI 10.1021/ja00767a075.
- M. S. Kharasch; E. K. Fields. Factors Determining the Course and Mechanisms of Grignard Reactions. IV. The Effect of Metallic Halides on the Reaction of Aryl Grignard Reagents and Organic Halides1. Journal of the American Chemical Society. 1941-09-01, s. 2316–2320. DOI 10.1021/ja01854a006.
- Jay K. Kochi; Masuhiko Tamura. Mechanism of the silver-catalyzed reaction of Grignard reagents with alkyl halides. Journal of the American Chemical Society. 1971, s. 1483–1485. DOI 10.1021/ja00735a028.
- Jay K. Kochi; Masuhiko Tamura. Alkylcopper(I) in the coupling of Grignard reagents with alkyl halides. Journal of the American Chemical Society. 1971-03-01, s. 1485–1487. DOI 10.1021/ja00735a029.
- Masuhiko Tamura; Jay K. Kochi. Vinylation of Grignard reagents. Catalysis by iron. Journal of the American Chemical Society. 1971-03-01, s. 1487–1489. DOI 10.1021/ja00735a030.
- Masaaki Yamamura; Ichiro Moritani; Shun-Ichi Murahashi. The reaction of σ-vinylpalladium complexes with alkyllithiums. Stereospecific syntheses of olefins from vinyl halides and alkyllithiums. Journal of Organometallic Chemistry. 1975-05-27, s. C39–C42. DOI 10.1016/S0022-328X(00)89636-9.
- Christiane E. I. Knappke; Axel Jacobi von Wangelin. 35 years of palladium-catalyzed cross-coupling with Grignard reagents: how far have we come?. Chemical Society Reviews. 2011, s. 4948–4962. DOI 10.1039/c1cs15137a. PMID 21811712.
- Xile Hu. Nickel-catalyzed cross coupling of non-activated alkyl halides: a mechanistic perspective. Chemical Science. 2011, s. 1867–1886. DOI 10.1039/c1sc00368b.
- Gavin D. Jones; Chris McFarland; Thomas J. Anderson; David A. Vicic. Analysis of key steps in the catalytic cross-coupling of alkyl electrophiles under Negishi-like conditions. Chemical Communications. 2005-01-01, s. 4211–4213. DOI 10.1039/b504996b. PMID 16100606.
- Anja C. Frisch; Matthias Beller. Catalysts for Cross-Coupling Reactions with Non-activated Alkyl Halides. Angewandte Chemie International Edition. 2005-01-21, s. 674–688. DOI 10.1002/anie.200461432. PMID 15657966.
- Michael E. Limmert; Amy H. Roy; John F. Hartwig. Kumada Coupling of Aryl and Vinyl Tosylates under Mild Conditions. The Journal of Organic Chemistry. 2005-11-01, s. 9364–9370. DOI 10.1021/jo051394l. PMID 16268609.
- Carl A. Busacca; Magnus C. Eriksson; Rita Fiaschi. Cross coupling of vinyl triflates and alkyl Grignard reagents catalyzed by nickel(0)-complexes. Tetrahedron Letters. 1999, s. 3101–3104. DOI 10.1016/S0040-4039(99)00439-6.
- Alena Rudolph; Mark Lautens. Secondary Alkyl Halides in Transition-Metal-Catalyzed Cross-Coupling Reactions. Angewandte Chemie International Edition. 2009-03-30, s. 2656–2670. DOI 10.1002/anie.200803611. PMID 19173365.
- Jun Terao; Hideyuki Watanabe; Aki Ikumi; Hitoshi Kuniyasu; Nobuaki Kambe. Nickel-Catalyzed Cross-Coupling Reaction of Grignard Reagents with Alkyl Halides and Tosylates: Remarkable Effect of 1,3-Butadienes. Journal of the American Chemical Society. 2002-04-01, s. 4222–4223. DOI 10.1021/ja025828v. PMID 11960446.
- Javier Adrio; Juan C. Carretero. Functionalized Grignard Reagents in Kumada Cross-Coupling Reactions. ChemCatChem. 2010-11-15, s. 1384–1386. DOI 10.1002/cctc.201000237.
- M. Kumada. Nickel and palladium complex catalyzed cross-coupling reactions of organometallic reagents with organic halides. Pure and Applied Chemistry. 1980-01-01, s. 669–679. DOI 10.1351/pac198052030669.
- Alois Fürstner; Andreas Leitner; María Méndez; Helga Krause. Iron-Catalyzed Cross-Coupling Reactions. Journal of the American Chemical Society. 2002-11-01, s. 13856–13863. DOI 10.1021/ja027190t. PMID 12431116.
- Tamio Hayashi, Akihiro Yamamoto, Masahiro Hojo, Kohei Kishi, Yoshihiko Ito, Eriko Nishioka, Hitoshi Miura, Kazunori Yanagi. Asymmetric synthesis catalyzed by chiral ferrocenylphosphine-transition metal complexes. Journal of Organometallic Chemistry. 1989, s. 129–139. DOI 10.1016/0022-328X(89)87280-8.
- Sha Lou; Gregory C. Fu. Nickel/Bis(oxazoline)-Catalyzed Asymmetric Kumada Reactions of Alkyl Electrophiles: Cross-Couplings of Racemic α-Bromoketones. Journal of the American Chemical Society. 2010, s. 1264–1266. DOI 10.1021/ja909689t. PMID 20050651.
- Yoshiharu Ikoma; Kazuhiko Ando; Yoshitake Naoi; Takeo Akiyama; Akira Sugimori. Halogen Selectivity in Nickel Salt-Catalyzed Cross-Coupling of Aryl Grignard Reagents with Bromochlorobenzenes a Novel Synthetic Method of Unsymmetrical Terphenyl. Synthetic Communications. 1991-02-01, s. 481–487. DOI 10.1080/00397919108016772.
- Modern Drug Synthesis. [s.l.]: John Wiley & Sons, 2010. Dostupné online. ISBN 978-0-470-52583-8. S. 153–154.
- Yen-Ju Cheng; Sheng-Hsiung Yang; Chain-Shu Hsu. Synthesis of Conjugated Polymers for Organic Solar Cell Applications. Chemical Reviews. 2009-11-11, s. 5868–5923. DOI 10.1021/cr900182s. PMID 19785455.
- Richard D. McCullough; Renae D. Lowe. Enhanced electrical conductivity in regioselectively synthesized poly(3-alkylthiophenes). Journal of the Chemical Society, Chemical Communications. 1992, s. 70. DOI 10.1039/C39920000070.
- Robert S. Loewe; Paul C. Ewbank; Jinsong Liu; Lei Zhai; Richard D. McCullough. Regioregular, Head-to-Tail Coupled Poly(3-alkylthiophenes) Made Easy by the GRIM Method: Investigation of the Reaction and the Origin of Regioselectivity. Macromolecules. 2001-06-01, s. 4324–4333. DOI 10.1021/ma001677+.