Konjugovaný systém
Jako konjugovaný systém se v chemii označuje systém propojených orbitalů p v molekule; ten obecně způsobuje snížení energie molekuly a nárůst její stability. Často se znázorňuje jako střídání jednoduchých a dvojných vazeb. Součástí systému mohou být volné elektronové páry, radikály nebo karbeniové ionty; struktura může být lineární i rozvětvená, acyklická, částečné i zcela cyklická. Pojem „konjugovaný systém“ poprvé použil roku 1899 německý chemik Johannes Thiele.[1]

Konjugace vzniká překryvem dvou orbitalů p (u přechodných kovů může jít i o orbitaly d) přes vazbu sigma.
V konjugovaném systému se nachází oblast s překryvem orbitalů p nebo d, která přechází přes mezilehlá místa, u kterých se na jednoduchých diagramem ukazuje, že se zde nenacházejí vazby pí. Dochází k delokalizaci π elektronů přes všechny propojené orbitaly p;[2] tyto elektrony nenáleží ke konkrétnímu atomu, ale spíše ke skupině atomů.
Největší konjugované systémy se nacházejí v grafenu, grafitu, vodivých polymerech a uhlíkových nanotrubicích.
Chemické vazby v konjugovaných systémech

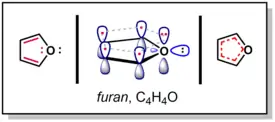
Konjugace nejčastěji spočívá ve střídání jednoduchých a dvojných vazeb, kdy má každý atom orbital p umístěný kolmo k rovině molekuly; za konjugovaný lze však považovat každý systém, ve kterém mají po sobě následující atomy volné orbitaly p; příkladem může být furan, který má pětičlenný kruh s dvojicí dvojných vazeb, které obklopují atom kyslíku. Na kyslíku se nacházejí dva volné elektronové páry, jeden se nachází v orbitalu p kolmém na rovinu molekuly, konjugace je udržována překryvem orbitalů obou vazebných atomů. Druhý elektronový pár zůstává v molekulové rovině a na konjugaci se nepodílí.
Obecně vzato se konjugace se může zúčastnit jakýkoliv sp2 nebo sp-hybridizovaný uhlík či heteroatom, i ty, které mají prázdné orbitaly nebo v nich mají volné elektronové páry, i když ty se nemusí vždy podílet na konjugaci. V molekule pyridinu se na ní, skrze dvojnou vazbu se sousedním uhlíkem, podílí i dusíkový atom a volný pár zůstává v rovině molekuly v hybridizovaném orbitalu sp2 a na konjugaci nemá vliv. Ke konjugaci je nutný překryv orbitalů a molekula tedy musí být zcela nebo téměř rovinná. Volné páry účastnící se konjugace se tak vyskytují v „čistých“ p orbitalech namísto hybridních orbitalů spn, ve kterých jsou elektronové páry, které konjugaci nevytváří.
Elektrony v konjugovaných π systémech jsou sdíleny sp2- a sp-hybridizovanými orbitaly, které se posílejí na překryvech; v důsledku toho se π elektrony v nich chovají jako jeden velký systém vazeb; často se popisují jako n-centrové k-elektronové vazby π, což se zobrazuje pomocí symbolů Π k
n . Například delokalizované elektrony v octanovém aniontu a v benzenu se nacházejí v Π 4
3 , respektive Π 6
6 systémech.
Stabilizační energie
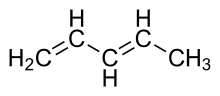
Odhady míry stabilizace vytvořené konjugovaným systémem jsou často nejisté. Energie stabilizace se nazývá rezonanční energie a je definována jako rozdíl skutečné energie sloučeniny a hypotetické sloučeniny se zcela lokalizovanými vazbami π odpovídajícími nejstabilnější rezonanční struktuře; tuto energii nelze změřit a přesná definice přijatelná pro většinu chemiků je nedosažitelná, lze však o ní vyslovit několik tvrzení. Stabilizace je obecně významnější u kationtových systémů než u neutrálních. U buta-1,3-dienu jde zhruba o aktivační energii rotace C2-C3, která má hodnotu přibližně 25 kJ/mol,[3] zatímco energetická bariéra rotace kolem dvojné vazby allylového kationtu v plynném skupenství je asi 160 kJ/mol.[4] Srovnáním afinity hydridového iontu k propylovému a allylovému kationtu, se započtením indukčních efektů, vychází 83 až 92 kJ/mol.[5]
I když je vliv stabilizace většinou poměrně slabý, tak u aromatických sloučenin může být výraznější. Odhady rezonanční energie benzenu se pohybují od 150 do 310 kJ/mol.[6]
Cyklické konjugované sloučeniny
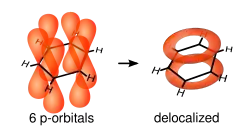

Cyklické sloučeniny mohou být částečně či zcela konjugované. Anuleny, plně konjugované monocyklické uhlovodíky, mohou být aromatické, nearomatické i antiaromatické.
Aromatické sloučeniny
Rovinné molekuly s konjugovaným systémem 4n + 2 (kde n je celé číslo) π elektronů jsou aromatické. Příkladem je benzen, který má 6 π elektronů, které s 12 σ elektrony vazeb C-C a 6 σ elektrony vazeb C-H vytváří termodynamicky a kineticky stabilní benzenový kruh; u benzenu se vyskytují dvě ekvivalentní konjugované Lewisovy struktury, které převažují (na celkové energii vazeb se podílí z více než 90 %).[7]
Skutečná elektronová struktura je kvantově mechanickou kombinací těchto variant, což vede k pozorovaným vlastnostem vazeb C–C, které délkou i energií tvoří přechod mezi jednoduchými a dvojnými vazbami. Na diagramu molekulových orbitalů se šest atomových orbitalů p spojuje v šest molekulových orbitalů. Tři z těchto orbitalů mají nižší energii než „čistý“ orbital p a jsou zaplněny šesti elektrony, zatímco tři destabilizované orbitaly protivazebné povahy zůstávají neobsazeny, což vede k termodynamické i kinetické stabilizaci.
Nearomatické a antoaromatické sloučeniny
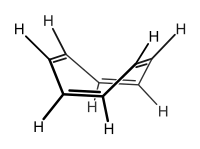
Ne všechny cyklické sloučeniny, ve kterých se střídají jednoduché a dvojné vazby, jsou aromatické; například cyklooktatetraen se obvykle vyskytuje v neplanárních konformacích. Jelikož do sebe p orbitaly nezapadají, tak jsou vazby π izolované a nikoliv konjugované. Nepřítomnost konjugace zamezuje antiaromaticitě molekuly, destabilizačnímu efektu objevujícímu se u molekul s 4n π (n = 0, 1, 2, ...) elektrony. Jelikož se zde dva elektrony nacházejí v degenerovaných nevazebných orbitalech, tak dochází k výraznému omezení termodynamické stabilizace. Tím dochází ke značnému nárůstu kinetické reaktivity látky. Jelikož je tento efekt velmi nepříznivý, tak cyklooktatetraen zaujímá nerovinnou konformaci a není aromatický, chová se tak jako alken. U derivátů cyklooktatetraenového dikationtu a dianiontu byl však experimentálně potvrzen rovinný tvar molekuly, což je v souladu s tím, že jde o stabilizované aromatické systémy s 6 či 10 π elektrony. Jelikož je antiaromaticita nevýhodnou vlastností, tak je známo jen několik takových látek, jako například cyklobutadien a cyklopentadienylový kationt.
Konjugované systémy u barviv
V konjugovaném π systému mohou elektrony pohltit fotony určitých frekvencí, které odpovídají vzdálenosti p orbitalů; podobně jako u antény. Obvykle platí, že čím je konjugovaný systém delší, tím delší je vlnová délka záření, které může pohltit.[8]
V molekulách mnoha barviv se nacházejí konjugované systémy elektronů, které jim díky absorpci viditelného světla dodávají výrazná zbarvení; například dlouhý konjugovaný uhlovodíkový řetězec u betakarotenu vede k oranžové barvě. Když elektron absorbuje foton, může se dostat na vyšší energetickou hladinu. Jednoduchý model energetických hladin lze vytvořit pomocí modelu potenciálové jámy délky L, která představuje pohyb π elektronu podél konjigovaného řetězce uhlíkových atomů. V tomto modelu nejmenší možná absorbovaná energie odpovídá rozdílu energie mezi nejvyšším obsazeným (HOMO) a nejnižším neobsazeným (LUMO) molekulovým orbitalem. V řetězci s n vazbami C=C nebo 2 n atomy uhlíku v základním stavu molekuly je 2 n π elektronů v n molekulových ornitalech, takže rozdíl energie má následující hodnotu:[9]

Protože se délka L s rostoucím počtem n vazeb C=C zvyšuje téměř lineárně, tak je energie ΔE absorbované při přechodu z HOMO do LUMO přibližně přímo úměrná 1/n; vlnová délka fotonu je tedy přibližně přímo úměrná n. I když je tento model značně zjednodušený, tak λ obecně se zvyšujícím se n (nebo L) u podobných molekul roste. Jako příklad je možné uvést vlnové délky absorbované v konjugovaných systémech v molekulách buta-1,3-dienu (217 nm), hexa-1,3,5-trienu (252 nm) a okta-1,3,5,7-tetraenu (304 nm).[10] K získání přesných výsledků je však třeba do modelu zahrnou také délky jednoduchých vazeb.[11]
K předpovídání absorpčních vlnových délek lze též použít Hückelovu metodu.
Mnoho přechodů elektronů v konjugovaných π systémech probíhá z vazebného do nevazebného molekulového orbitalu (tedy z π do π*), ovšem elektrony z nevazebných volných párů mohou rovněž přejít do π systému MO. K přechodu elektronu z HOMO do LUMO dojde tehdy, když takový přechod „dovolují“ určitá pravidla. Konjugované systémy s méně než osmi dvojnými vazbami absorbují pouze ultrafialové záření a jsou neviditelné. S každou další dvojnou vazbou se prodlužuje vlnová délka (a tedy snižuje energie) absorbovaných fotonů a barva sloučeniny se posouvá od žluté k červené. U látek s modrým či zeleným zabarvením obvykle není absorpce založená pouze na konjugovaném systému.
Pohlcování elektromagnetického záření v ultrafialové a viditelné oblasti se využívá v ultrafialovo-viditelné spektroskopii a je základním principem ve fotochemii.
K často využívaným druhům konjugovaných systémů patří diazosloučeniny, azosloučeniny a ftalokyaniny.
Ftalokyaniny
Konjugované systémy se nevyznačují pouze nízkou excitační energií, ale rovněž i snadným přijetím či odevzdáním elektronů. Ftalokyaniny, jako jsou ftalokyaninová modř BN a ftalokyaninová zeleň G, mají často ve svých molekulách atomy přechodných kovů a vyměňují si elektrony s jejich komplexovanými ionty, čímž snadno dochází ke změně oxidačního čísla.

Porfyriny a podobné sloučeniny
Porfyriny mají konjugované cyklické molekulové systémy, které jsou součástí řady enzymů. Porfyriny, jako ligandy, tvoří mnoho komplexů s ionty kovů; například železo v hemoglobinu dodává krvi červenou barvu. Další struktura nazývaná chlorin podobně vytváří komplex s hořečnatými ionty; vyskytuje se v chlorofylech, kterým dodává zelenou barvu. Korin je skupina, která se komplexuje s kobaltnatými kationty za vzniku kobalaminových molekul, například vitaminu B12, který je červený. Korinová jednotka má šest konjugovaných dvojných vazeb, ovšem není konjugovaná po celém makrocyklickém kruhu.
 |  |  |
| Hemová skupina hemoglobinu | chlorinová část molekuly chlorofylu a. Zeleně je označena funkční skupina, která se u různých druhů chlorofylů liší. | Struktura kobalaminu obsahuje korinový makrocyklus. |
Chromofory
Konjugované systémy tvoří základy chromoforů, což jsou části molekul, které absorbují světlo a dodávají jim tak barvu. Chromofory často obsahují řetězce konjugovaných vazeb a/nebo cyklů, často aromatických, v nichž mohou být přítomny vazby C–C, C=C, C=O a N=N.

Konjugované chromofory jsou součástí řady organických sloučenin jako jsou látky přirozeně obsažené v potravinách (lykopen a antokyanidiny), fotoreceptory, azobarviva a některá léčiva, mimo jiné tato:
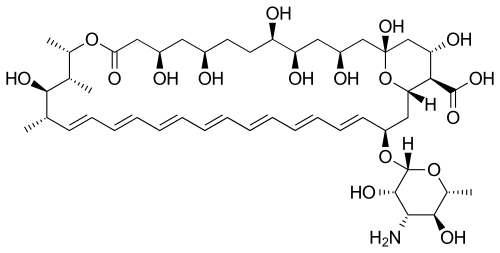 Antimykotikum amfotericin B má konjugovaný systém sedmi dvojných vazeb, který funguje jako chromofor silně absorbující v ultrafialovém a viditelném spektru, díky čemuž je zbarven do žluta.
Antimykotikum amfotericin B má konjugovaný systém sedmi dvojných vazeb, který funguje jako chromofor silně absorbující v ultrafialovém a viditelném spektru, díky čemuž je zbarven do žluta.
Odkazy
Související články
- Rezonanční struktura
- Hyperkonjugace
- Zkřížená konjugace
- Polyeny
Reference
V tomto článku byl použit překlad textu z článku Conjugated system na anglické Wikipedii.
- Johannes Thiele. Zur Kenntnis der ungesättigten Verbindungen. Justus Liebigs Annalen der Chemie. 1899, s. 90. Dostupné online. (německy)
- March Jerry; (1985). Advanced Organic Chemistry reactions, mechanisms and structure (3rd ed.). New York: John Wiley & Sons, inc. ISBN 0-471-85472-7
- David Feller; Norman C. Craig. High Level ab Initio Energies and Structures for the Rotamers of 1,3-Butadiene. Journal of Physical Chemistry A. 2. 5. 2009, s. 1601–1607. PMID 19199679. (anglicky)
- Alberto Gobbi; Gernot Frenking. High Level ab Initio Energies and Structures for the Rotamers of 1,3-Butadiene. Journal of the American Chemical Society. 1. 10. 1994, s. 9275–9286. ISSN 0002-7863. (anglicky)
- Josiah B. Barbour; Joel M. Karty. Resonance Energies of the Allyl Cation and Allyl Anion: Contribution by Resonance and Inductive Effects toward the Acidity and Hydride Abstraction Enthalpy of Propene. Journal of Organic Chemistry. 14. 1. 2004, s. 648–654. PMID 14750787. (anglicky)
- Frank Albert Cotton. Chemical applications of group theory. New York: Wiley, 1990. (3). Dostupné online. ISBN 978-0471510949. OCLC 19975337 (anglicky)
- Zahid Rashid; Joop H. van Lenthe. Generation of Kekulé valence structures and the corresponding valence bond wave function. Journal of Computational Chemistry. 3. 2011, s. 696–708. ISSN 1096-987X. PMID 20941739. (anglicky)
- Mark Lipton. Purdue: Chem 26505: Organic Chemistry I (Lipton). [s.l.]: Purdue University, 31. 1. 2017. (LibreTexts). Dostupné online. Kapitola Chapter 1. Electronic Structure and Chemical Bonding. (anglicky)
- P. Atkins and J. de Paula Physical Chemistry (8th ed., W.H.Freeman 2006), p.281 ISBN 0-7167-8759-8
- Atkins and de Paula p.398
- Jochen Autschbach. Why the Particle-in-a-Box Model Works Well for Cyanine Dyes but Not for Conjugated Polyenes. Journal of Chemical Education. 11. 2007, s. 1840. ISSN 0021-9584. (anglicky)