Wackerův proces
Wackerův proces, také nazývaný Hoechstův-Wackerův proces, je oxidace ethenu na acetaldehyd za přítomnosti chloridu palladnatého jako katalyzátoru. Jedná se o jeden z prvních známých případů homogenní katalýzy organických sloučenin palladia využívaných v průmyslu.[1]
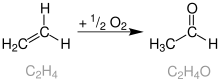
Historie
Tuto reakci poprvé popsali J. Smidt et al.[2][3][4]
Rozvoj reakce později nazývané Wackerův proces začal v roce 1956.[5] V této době se mnoho průmyslově využívaných sloučenin vyrábělo z acetylenu, jehož prekurzorem byl nákladně vyráběný karbid vápenatý. Později byla vyvinuta reakce ethenu s kyslíkem katalyzovaná palladiem na vrstvě uhlíku, původně určená na výrobu ethylenoxidu, ovšem zjistilo se, že produktem je acetaldehyd. Další výzkum této reakce vedl v roce 1957 k patentu popisujícímu reakci v plynné fázi za použití heterogenního katalyzátoru.[6] Heterogenní proces se ukázal jako nevhodný, protože docházelo k deaktivaci katalyzátoru, byl tak roku 1958 nahrazen homogenní katalýzou ve vodném prostředí. Potíže s agresivitou katalytického roztoku byly vyřešeny použitím titanu na výrobu reaktorů a pump. Výroba tímto způsobem byla zahájena v roce 1960.
Mechanismus
Mechanismus průmyslového Wackerova procesu (oxidace alkenů za přítomnosti chloridu palladnatého) byl po několik desetiletí významným předmětem výzkumu; některé detaily stále nejsou jasné. Níže je popsán moderní popis mechanismu:
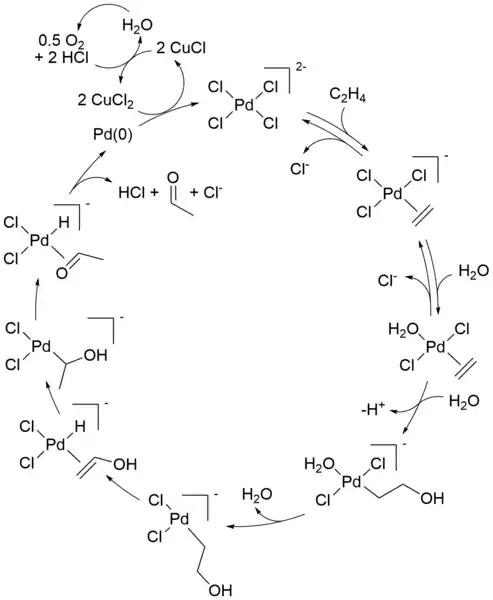
Úvodní stechiometrickou reakci popsal C. Phillips.[8][9] Lze ji vyjádřit následující rovnicí:
- [PdCl4]2 − + C2H4 + H2O → CH3CHO + Pd + 2 HCl + 2 Cl−
Po této přeměně následují reakce, které vedou k obnově palladnatého katalyzátoru:
- Pd + 2 CuCl2 + 2 Cl − → [PdCl4]2− + 2 CuCl
- 2 CuCl + ½ O2 + 2 HCl → 2 CuCl2 + H2O
Při reakci se spotřebovávají pouze alken a kyslík. Bez chloridu měďnatého jako oxidačního činidla by se sráželo kovové palladium (vzniklé beta-hydridovou eliminací palladnaté sloučeniny v posledním kroku), čímž by se reakce zastavila po jednom cyklu; tato stechiometrická reakce byla objevena v roce 1894. Vzniklý chlorid měďný může být oxidován vzduchem, čistým kyslíkem i řadou dalších látek za přeměny zpět na CuCl2 což umožňuje pokračování katalytického cyklu.
Mechanistické studie
První mechanistické studie z 60. let 20. století ukázaly na několik klíčových míst:[7][10]
- Při reakci nebyly pozorovány žádné H/D výměny. Experimenty s C2D4 ve vodě vznikal CD3CDO a u C2H4 v D2O se tvořil CH3CHO. Součástí mechanismu tedy není keto-enolová tautomerie.
- Při použití zcela deuterovaných reaktantů se neobjevovaly téměř žádné kinetické izotopové efekty, což ukazuje, že přenos hydridu není krokem určujícím rychlost reakce.
- Výrazný izotopový efekt s C2H2D2 naznačuje, že krok určující rychlost se odehrává před tvorbou acetaldehydu.
- Vysoké koncentrace chloridů vyvolávají tvorbu dalšího produktu, jímž je chlorhydrin.
Mnoho mechanistických studií zaměřených na Wackerův proces se zabývalo způsobem vzniku vazby C-O v hydroxypalladačním kroku. Bylo zjištěno, že koordinovaný hydroxid reaguje s ethenovým ligandem, a to vnitřním (syn-) mechanismem.[11] Později stereochemické studie podpořily anti-adici, jejímž prostřednictvím se volný hydroxid váže na ethen.[12][13][14] Podmínky těchto experimentů byly značně odlišné od průmyslových. Při ostatních studiích se používaly reakční podmínky odpovídající průmyslovému Wackerovu procesu (s výjimkou vysoké koncentrace chloridu palladnatého a měďnatého) a také se tvořily produkty naznačující průběh nukleofilního ataku anti-adičně.[15]
Byly taktéž udělány kinetické studie provedené u izotopicky substituovaných allylalkoholů za průmyslových podmínek (s nízkými koncentracemi chloridů).[16][17] Podle nich nukleofilní atak probíhá pomalu, přestože mechanismy navržené podle předchozích stereochemických studiích ukazovaly na rychlý průběh této části procesu.
Dalšími stereochemickými studiemi bylo zjištěno, že se objevují oba mechanismy a jejich podíly závisí na koncentracích chloridů.[18][19] Výsledky těchto studií jsou ovšem sporné, protože allylalkoholy mohou být náchylné k izomerizacím a tvorba rozdílných stereoizomerů může být způsobena těmito vedlejšími reakcemi a ne samotným Wackerovým procesem.
Experimentální důkazy tak ukazují na syn-adice při nízkých koncentracích chloridů (pod 1 mol/l, odpovídající průmyslovým podmínkám), zatímco anti-adice převažují za vysokých koncentrací chloridů (nad 3 mol/l), což je pravděpodobně způsobeno tím, že chloridové ionty způsobují nasycení katalyzátoru a inhibují tak mechanismus založený na přenosu ve vnější sféře. Přesný průběh reakce a příčina přechodů mezi těmito mechanismy ovšem dosud nejsou známy.
Obtíže při zkoumání mechanismu Wackerova procesu způsobuje také nejasná funkce chloridu měďnatého. Podle většiny teorií nemá měď vliv na mechanismus oxidace alkenů, ovšem experimenty, jež provedli H. Stangl a R. Jira[20] ukázaly, že tvorba chlorohydrinu je závislá na koncentraci CuCl2. T. Hosokawa zjistil,[21] vzniká krystalizovaný produkt obsahující chlorid měďnatý, což naznačuje určitý jeho vliv na oxidaci. Později bylo ab initio studií, kterou provedli A. Comas-Vives et al,[22] že za nepřítomnosti tohoto kokatalyzátoru převažuje anti-adice; což bylo později potvrzeno obdobnými experimenty B. J. Andersona a J. A. Sigmana.[23] Za nepřítomnosti mědi se objevuje jiná reakční kinetika, nezávislá na protonech, takže na průběh reakce mohou mít vliv i malá množství kokatalyzátoru.
Další důležitou součástí Wackerova procesu je přesun vodíku z kyslíkového atomu na chlorid a tvorba dvojné vazby C-O. Tento krok nejspíše probíhá přes beta-hydridovou eliminaci s čtyřčlenným cyklickým meziproduktem:
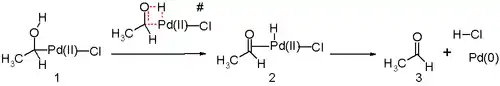
Ve studiích na základě metod výpočetní chemie bylo zjištěno, že příslušný přechodný stav by měl být nevýhodný a převažuje pravděpodobně mechanismus založený na redukční eliminaci. Katalyzátorem jsou molekuly vody přítomné v roztoku.
Průmyslové provedení
Při průmyslové výrobě acetaldehydu se používají dva postupy: jednofázový a dvoufázový.
Jednofázový proces
Při jednofázovém procesu se ethen a kyslík současně dodávají do reaktoru při teplotě okolo 130 °C a tlaku 400 kPa.[24] Katalyzátorem je vodný roztok PdCl2 a CuCl2. Acetaldehyd se přečišťuje extrakční a následně frakční destilací. Extrakční destilace s vodou oddělí vedlejší produkty s nižšími teplotami varu, než má acetaldehyd (například chlormethan, chlorethan a oxid uhličitý) v horní části nádoby, zatímco voda a další výševroucí látky, jako kyselina octová, krotonaldehyd a chloracetaldehydy, se společně s acetaldehydem oddělí v dolní části.[24] Vzhledem k žíravosti katalyzátoru je reaktor chráněn vrstvou keramického materiálu a potrubí je tvořené titanem.
Dvoufázový proces
Dvoufázový proces se skládá ze dvou samostatně prováděných kroků. Na rozdíl od jednofázového zde lze použít oxidaci vzduchem. Ethen prochází reaktorem společně s katalyzátorem za teploty 105–110 °C, přičemž tlak činí 900–1000 kPa.[24] Roztok katalyzátoru obsahující acetaldehyd se oddělí rovnovážnou destilací. Katalyzátor se v oxidačním reaktoru při 1000 kPa oxiduje vzduchem. Roztok oxidovaného katalyzátoru se následně vrátí do reaktoru. Kyslík se spotřebuje úplně a zbylý vzduch funguje jako netečný plyn. Směs par acetaldehydu a vody se prekoncentruje na 60–90 % acetaldehydu pomocí reakčního tepla a oddělená voda putuje zpět do prvního reaktoru, kde udržuje koncentraci katalyzátoru. Poté se provede dvoufázová destilace surového acetaldehydu; v první části se oddělí nízkovroucí vedlejší produkty, jako jsou chlormethan, chlorethan a oxid uhličitý. Ve druhé dojde k oddělení vody a vedlejších produktů s vyššími teplotami varu, jako jsou chlorované deriváty acetaldehydu a kyselina octová, čímž se vytvoří čistý acetaldehyd.[24]
V obou variantách je výtěžnost acetaldehydu okolo 95%[24] a i nákladnost je v podstatě stejná. Výhoda použití rozpuštěných plynů v dvoufázovém postupu je vyvážena vyššími vstupními náklady. Při obou postupech vznikají chlorované uhlovodíky, chlorované acetaldehydy a kyselina octová. Výběr způsobu výroby závisí na surových materiálech, nákladech na energie a na dostupnosti nepříliš nákladného kyslíku.
Obecně ze 100 dílů ethenu vzniká:
- 95 dílů acetaldehydu
- 1,9 dílů chloraldehydů
- 1,1 dílů nevyužitého ethenu
- 0,8 dílů oxidu uhličitého
- 0,7 dílů kyseliny octové
- 0,1 dílů chlormethanu
- 0,1 dílů ethylchloridu
- 0,3 dílů ethanu, methanu, krotonaldehydu a dalších vedlejších produktů.
 Diagram jednofázového Wackerova procesu
Diagram jednofázového Wackerova procesu Diagram dvoufázového Wackerova procesu
Diagram dvoufázového Wackerova procesu
Cudžiova-Wackerova oxidace
Objev Wackerova procesu spustil výzkum využitelnosti reakce u složitějších koncových alkenů. Cudžiova-Wackerova oxidace je přeměna takových alkenů, katalyzovaná palladnatými sloučeninami, na karbonylové sloučeniny. William H. Clement a Charles M. Selwitz[25] použití vodného roztoku dimethylformamidu (DMF) jako rozpouštědla umožňuje oxidovat dodec-1-en na dodekan-2-on, čímž byla vyřešena nerozpustnost vyšších alkenů ve vodě. Darryl R. Fahey[26] použil 3-methylsulfolan namísto DMF a a navýšil výtěžnost oxidace 3,3-dimethylbut-1-enu. O dva roky později Džiró Cudži[27] využil tento postup k selektivním oxidacím koncových alkenů s několika různými funkčními skupinami a předvedl jeho využitelnost při přípravě komplexních substrátů.[28]
Další průzkum této reakce vedl k vývoji různých katalytických systémů za účelem provedení selektivních reakcí a provádění mezimolekulárních i vnitromolekulárních oxidací pomocí nukleofilů neobsahujících vodu.
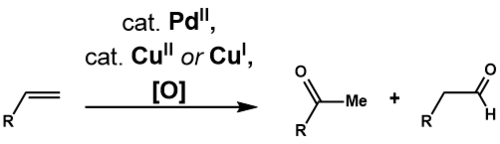
Markovnikovovská adice
Cudžiova-Wackerova oxidace oxiduje alken na odpovídající methylketon. Téměř stejný jako u Wackerova procesu je navržený katalytický cyklus[29] začínající komplexací PdCl2 s dvěma chloridovými anionty za vzniku PdCl4, který poté vymění dva chloridové ligandy za vodu a alken, čímž se vytvoří komplex Pd(Cl2)(H2O)(alken). Molekula vody pak regioselektivně atakuje alken ve vnější sféře v souladu s Markovnikovovým pravidlem, čímž vznikne termodynamicky stabilní Pd(Cl2)(OH)(-CH2-CHOH-R). Odštěpení chloridového ligandu a jeho napojení na tříkoordinovaný komplex palladia spustí beta-hydridovou eliminaci, po níž dojde k 1,2-inserci hydridu za vzniku Pd(Cl2)(OH)(-CHOHR-CH3). Tento komplex projde beta-hydridovou eliminací, jejímž produktem je keton, a poté redukční eliminací za vzniku HCl, vody a Pd0. Toto palladium je nakonec zpětně oxidováno na PdCl2 pomocí dvou ekvivalentů CuCl2, u kterých se oxidační číslo II obnoví působením O2.
Oxidací koncových alkenů většinou vznikají markovnikovovské produkty, když substrát upřednostňuje aldehyd, lze pomocí jiných ligandů také dosáhnout markovnikovovské regioselektivity. Použitím sparteinového ligandu[30] vede k upřednostnění nukleopaladace na koncovém uhlíku, protože se tím minimalizují sterické interakce mezi palladnatým komplexem a substrátem. Palladnatý katalyzátor ligovaný quinoxem lze použít k upřednostnění tvorby ketonu, pokud substrát obsahuje řídicí skupinu.[31]
Když se takový substrát naváže na Pd(quinox)(OOtBu), tak je komplex koordinačně nasycen, což zabraňuje navázání řídicí skupiny a vede k tvorbě markovnikovovského produktu. Účinky tohoto ligandu jsou přičítány jeho elektronovým vlastnostem, kde se aniontový TBHP váže především na pozici trans vůči oxazolinu a alken do polohy trans vzhledemk chinolinu.[32]
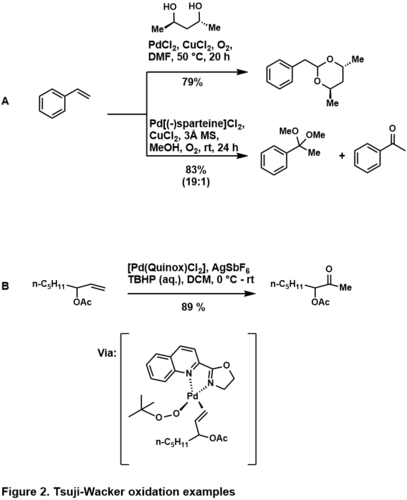
Protimarkovnikovovská adice
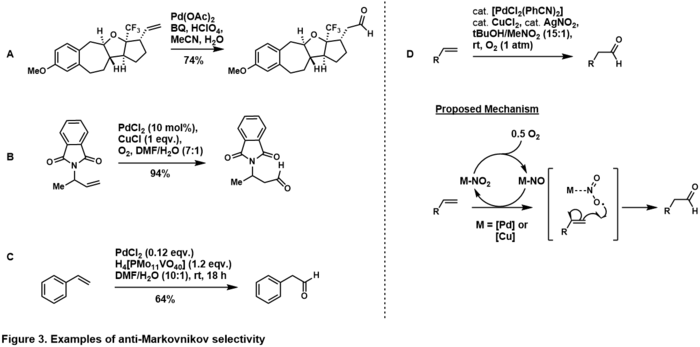
Protimarkovnikovovskou selektivitu vůči aldehydu je možné vytvořit úpravou stereoelektronových vlastností substrátu.[33] Umístěním řídicí skupiny na homoallylové[34] a allylové pozice[35] koncového alkenu dojde k kvorbě protimarkovnikovského aldehydu, což naznačuje, že se řídicí skupina v katalytickém cyklu chelatuje na komplex palladia takovým způsobem, že když voda atakuje protimarkovnikovský uhlík, tak je vzniklý palladocyklus termodynamicky stabilnější. Protimarkovnikovská selektivita se rovněž objevuje u styrenylových substrátů,[36] pravděpodobně přes η4-Pd-styrenový komplex po protimarkovnikovovském ataku molekulou vody. Další případy protimarkovnikovovských Cudžiových-Wackerových oxidací řízených substráty popsali I. N. N. Namboothiri,[37] Ben L. Feringa,[33] a Jacques Muzart.[38]
Robert H. Grubbs et al. provedli protimarkovnikovskou oxidaci stereoelektronicky nezatížených koncových alkenů s použitím systému založeného na palladiu a dusitanech;[39] při použití tohoto systému se alken oxiduje na aldehyd ve vysoce selektivní reakci řízené katalyzátorem. Mechanismus je předmětem výzkumu, objevují se ovšem důkazy,[37] že v průběhu reakce se dusitanový radikál navazuje na koncový uhlík za vzniku termodynamicky stabilního sekundárního radikálu.
Tento postup byl rozšířen na složitější alkeny.[40][41]
Kyslíkaté nukleofily
Mezimolekulové oxidace alkenů pomocí alkoholových nukleofilů obvykle vedou k tvorbě ketalů, při použití karboxylových kyselin vznikají vinyl- nebo allylkarboxyláty. U diolů se reakcemi s jednoduchými alkeny vytváří ketaly a u alkenů se skupinami odtahujícími elektrony většinou vznikají acetaly.[42]
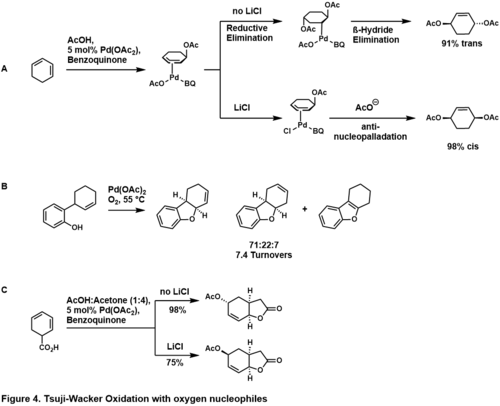
Mezimolekulární oxidace dienů karboxylovými nebo alkoholovými donory vedou k 1,4-adicím. U cyklohexadienu je stereochemie produktu závislá na koncentraci chlorid lithný.[43]
Při těchto reakcích se nejprve nukleopalladací dienu acetátovým nukleofilem vytváří Pd(OAc)(benzochinon)(allyl)ový komplex. Nepřítomnost LiCl vyvolává redukční eliminaci ve vnitřní sféře za tvorby trans-1,4-aduktu. Pokud se do rekční směsi přidá LiCl, dojde jeho účinkem k odstranění acetátu chloridem, který má vyšší vazebnou afinitu, vynucující atak acetátu v poloze anti vůči palladiu, a řídí reakci směrem k cis-1,4-aduktu. Následně proběhne oxidační cykloadice, kdy se 2-(2-cyklohexenyl)fenol cyklizuje na odpovídající dihydrobenzofuran;[44] kyselina 1-cyklohexadienoctová se za přítomnosti kyseliny octové mění na příslušný laktonacetátový 1,4 adukt,[45] cis/trans selektivitu lze ovládat přidáním LiCl.
Dusíkaté nukleofily
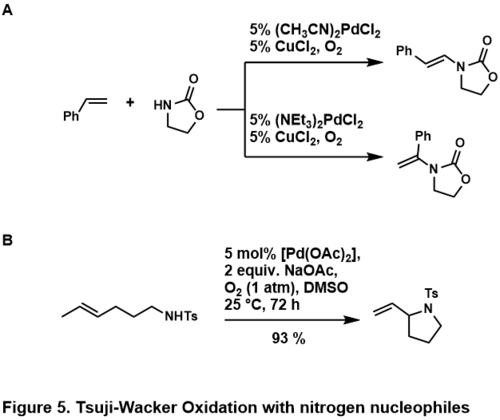
Oxidační aminace alkenů se obvykle provádějí pomocí amidů nebo imidů; aminy by byly v kyselém prostředí protonovány nebo by se na kov vázaly příliš silně, než aby se mohl projevit účinek katalyzátoru.[42] Tyto dusíkaté nukleofily mohou vstupovat do mezimolekulárních i vnitromolekulárních reakcí; několik příkladů je zobrazeno níže.[46][47]
Reference
V tomto článku byl použit překlad textu z článku Wacker process na anglické Wikipedii.
- Elschenbroich, C. "Organometallics" (2006) Wiley-VCH: Weinheim. ISBN 978-3-527-29390-2
- J. Smidt, W. Hafner, R. Jira, J. Sedlmeier, R. Sieber, R. Rüttinger, and H. Kojer, Angewandte Chemie, 1959, 71, 176–182. DOI:10.1002/ange.19590710503
- W. Hafner, R. Jira, J. Sedlmeier, and J. Smidt, Chem. Ber., 1962, 95, 1575–1581.
- J. Smidt, W. Hafner, R. Jira, R. Sieber, J. Sedlmeier, and A. Sabel, Angew. Chem. Int. Ed. Engl., 1962, 1, 80–88.
- Acetaldehyde from Ethylene — A Retrospective on the Discovery of the Wacker Process Reinhard Jira Angewandte Chemie International Edition 2009, 48, 9034–9037 DOI:10.1002/anie.200903992
- J. Smidt, W. Hafner, J. Sedlmeier, R. Jira, R. Rottinger (Cons. f.elektrochem.Ind.), DE 1 049 845, 1959, Anm. 04.01.1957.
- J. A. Keith; P. M. Henry. The Mechanism of the Wacker Reaction: A Tale of Two Hydroxypalladations. Angewandte Chemie International Edition. 2009, s. 9038–9049. DOI 10.1002/anie.200902194. PMID 19834921.
- F. C. Phillips, Am. Chem. J., 1894, 16, 255–277.
- F. C. Phillips, Zeitschrift für anorganische und allgemeine Chemie, 1894, 6, 213–228.
- Henry, Patrick M. In Handbook of Organopalladium Chemistry for Organic Synthesis; Negishi, E., Ed.; Wiley & Sons: New York, 2002; p 2119. ISBN 0-471-31506-0
- P. M. Henry, Journal of the American Chemical Society, 1964, 86, 3246–3250.
- James, D.E., Stille, J.K. Journal of Organometallic Chemistry, 1976, 108, 401. DOI:10.1021/ja00423a028
- Stille, J.K., Divakarumi, R.J., Journal of Organometallic Chemistry, 1979, 169, 239;
- James, D.E., Hines, L.F., Stille, J.K. Journal of the American Chemical Society, 1976, 98, 1806 DOI:10.1021/ja00423a027
- Bäckvall, J.E., Akermark, B., Ljunggren, S.O., Journal of the American Chemical Society, 1979, 101, 2411. DOI:10.1021/ja00503a029
- Zaw, K., Lautens, M. and Henry P.M. Organometallics, 1985, 4, 1286–1296
- Wan W.K., Zaw K., and Henry P.M. Organometallics, 1988, 7, 1677–1683
- Francis, J.W., Henry, P.M. Organometallics, 1991, 10, 3498. DOI:10.1021/om00056a019
- Francis, J. W., Henry, P. M. Organometallics, 1992, 11, 2832.DOI:10.1021/om00044a024
- H. Stangl and R. Jira, Tetrahedron Letters, 1970, 11, 3589–3592
- T. Hosokawa, T. Nomura, S.-I. Murahashi, Journal of Organometallic Chemistry, 1998, 551, 387–389
- Comas-Vives, A., Stirling, A., Ujaque, G., Lledós, A., Chemistry—A European Journal, 2010, 16, 8738–8747.DOI:10.1002/chem.200903522
- Anderson, B. J., Keith, J. A., Sigman, M. S., Journal of the American Chemical Society, 2010, 132, 11872-11874
- Šablona:Ullmann
- William H. Clement; Charles M. Selwitz. Improved Procedures for Converting Higher α-Olefins to Methyl Ketones with Palladium Chloride. The Journal of Organic Chemistry. 1964, s. 241–243. ISSN 0022-3263. DOI 10.1021/jo01024a517.
- Darryl R. Fahey; Ernest A. Zeuch. Aqueous sulfolane as solvent for rapid oxidation of higher .alpha.-olefins to ketones using palladium chloride. The Journal of Organic Chemistry. 1974, s. 3276–3277. ISSN 0022-3263. DOI 10.1021/jo00936a023.
- Jiro Tsuji; Isao Shimizu; Keiji Yamamoto. Convenient general synthetic method for 1,4- and 1,5-diketones by palladium catalyzed oxidation of α-allyl and α-3-butenyl ketones. Tetrahedron Letters. 1976, s. 2975–2976. ISSN 0040-4039. DOI 10.1016/s0040-4039(01)85504-0.
- Jiro Tsuji. Synthetic Applications of the Palladium-Catalyzed Oxidation of Olefins to Ketones. Synthesis. 1984, s. 369–384. ISSN 0039-7881. DOI 10.1055/s-1984-30848.
- Laszlo Kurti; Barbara Czako. Strategic Applications of named Reactions in Organic Synthesis. [s.l.]: Elsevier Academic Press, 2005. ISBN 978-0-12-429785-2. S. 474.
- Amy M. Balija; Kara J. Stowers; Mitchell J. Schultz; Matthew S. Sigman. Pd(II)-Catalyzed Conversion of Styrene Derivatives to Acetals: Impact of (−)-Sparteine on Regioselectivity. Organic Letters. 2006, s. 1121–1124. ISSN 1523-7060. DOI 10.1021/ol053110p. PMID 16524283.
- Brian W. Michel; Andrew M. Camelio; Candace N. Cornell; Matthew S. Sigman. A General and Efficient Catalyst System for a Wacker-Type Oxidation Using TBHP as the Terminal Oxidant: Application to Classically Challenging Substrates. Journal of the American Chemical Society. 2009-05-06, s. 6076–6077. ISSN 0002-7863. DOI 10.1021/ja901212h. PMID 19364100.
- Brian W. Michel; Laura D. Steffens; Matthew S. Sigman. On the Mechanism of the Palladium-Catalyzed tert -Butylhydroperoxide-Mediated Wacker-Type Oxidation of Alkenes Using Quinoline-2-Oxazoline Ligands. Journal of the American Chemical Society. 2011, s. 8317–8325. ISSN 0002-7863. DOI 10.1021/ja2017043. PMID 21553838.
- Jia Jia Dong; Wesley R. Browne; Ben L. Feringa. Palladium-Catalyzed anti-Markovnikov Oxidation of Terminal Alkenes. Angewandte Chemie International Edition. 2014-11-03, s. 734–744. ISSN 1433-7851. DOI 10.1002/anie.201404856. PMID 25367376.
- D. G. Miller; Danial D. M. Wayner. Improved method for the Wacker oxidation of cyclic and internal olefins. The Journal of Organic Chemistry. 1990, s. 2924–2927. ISSN 0022-3263. DOI 10.1021/jo00296a067.
- Roland Stragies; Siegfried Blechert. Enantioselective Synthesis of Tetraponerines by Pd- and Ru-Catalyzed Domino Reactions. Journal of the American Chemical Society. 2000, s. 9584–9591. ISSN 0002-7863. DOI 10.1021/ja001688i.
- Joseph A. Wright; Matthew J. Gaunt; Jonathan B. Spencer. Novel Anti-Markovnikov Regioselectivity in the Wacker Reaction of Styrenes. Chemistry - A European Journal. 2006-01-11, s. 949–955. ISSN 0947-6539. DOI 10.1002/chem.200400644. PMID 16144020.
- Thekke Veettil Baiju; Edmond Gravel; Eric Doris; Irishi N .N. Namboothiri. Recent developments in Tsuji-Wacker oxidation. Tetrahedron Letters. 2016, s. 3993–4000. ISSN 0040-4039. DOI 10.1002/chem.200400644. PMID 16144020.
- Jacques Muzart. Aldehydes from Pd-catalysed oxidation of terminal olefins. Tetrahedron. 2007, s. 7505–7521. ISSN 0040-4020. DOI 10.1016/j.tet.2007.04.001.
- Zachary K. Wickens; Bill Morandi; Robert H. Grubbs. Aldehyde-Selective Wacker-Type Oxidation of Unbiased Alkenes Enabled by a Nitrite Co-Catalyst. Angewandte Chemie International Edition. 2013-09-13, s. 11257–11260. Dostupné online. ISSN 1433-7851. DOI 10.1002/anie.201306756. PMID 24039135.
- Zachary K. Wickens; Kacper Skakuj; Bill Morandi; Robert H. Grubbs. Catalyst-Controlled Wacker-Type Oxidation: Facile Access to Functionalized Aldehydes. Journal of the American Chemical Society. 2014-01-13, s. 890–893. Dostupné online. ISSN 0002-7863. DOI 10.1021/ja411749k. PMID 24410719.
- Kelly E. Kim; Jiaming Li; Robert H. Grubbs; Brian M. Stoltz. Catalytic Anti-Markovnikov Transformations of Hindered Terminal Alkenes Enabled by Aldehyde-Selective Wacker-Type Oxidation. Journal of the American Chemical Society. 2016-09-30, s. 13179–13182. Dostupné online. ISSN 0002-7863. DOI 10.1021/jacs.6b08788. PMID 27670712.
- John F. Hartwig. Organotransition Metal Chemistry: From Bonding to Catalysis. [s.l.]: University Science Books, 2010. ISBN 978-1-891389-53-5. S. 717–734.
- Jan E. Baeckvall; Styrbjoern E. Bystroem; Ruth E. Nordberg. Stereo- and regioselective palladium-catalyzed 1,4-diacetoxylation of 1,3-dienes. The Journal of Organic Chemistry. 1980, s. 4619–4631. Dostupné online. ISSN 0022-3263. DOI 10.1021/jo00198a010.
- Takahiro Hosokawa; Shyogo Miyagi; Shunichi Murahashi; Akio Sonoda. Oxidative cyclization of 2-allylphenols by palladium(II) acetate. Changes in product distribution. The Journal of Organic Chemistry. 1978, s. 2752–2757. ISSN 0022-3263. DOI 10.1021/jo00408a004.
- Jan E. Baeckvall; Kenneth L. Granberg; Pher G. Andersson; Roberto Gatti; Adolf Gogoll. Stereocontrolled lactonization reactions via palladium-catalyzed 1,4-addition to conjugated dienes. The Journal of Organic Chemistry. 1993, s. 5445–5451. ISSN 0022-3263. DOI 10.1021/jo00072a029.
- Vitaliy I. Timokhin; Shannon S. Stahl. Brønsted Base-Modulated Regioselectivity in the Aerobic Oxidative Amination of Styrene Catalyzed by Palladium. Journal of the American Chemical Society. 2005, s. 17888–17893. ISSN 0002-7863. DOI 10.1021/ja0562806. PMID 16351120.
- Richard C. Larock; Timothy R. Hightower; Lisa A. Hasvold; Karl P. Peterson. Palladium(II)-Catalyzed Cyclization of Olefinic Tosylamides. The Journal of Organic Chemistry. 1996, s. 3584–3585. ISSN 0022-3263. DOI 10.1021/jo952088i. PMID 11667199.