Rhodocen
Rhodocen je organická sloučenina rhodia se vzorcem [Rh(C5H5)2]. Molekula se skládá z atomu rhodia navázaného mezi dva aromatické cyklopentadienylové cykly. Tato organokovová sloučenina obsahuje haptické kovalentní vazby rhodium–uhlík.[1] Radikál [Rh(C5H5)2] lze získat zahřátím nad 150 °C nebo zachycením pomocí chlazení kapalným dusíkem na −196 °C. Při pokojové teplotě se dvojice těchto radikálů spojí přes cyklopentadienylové cykly za vzniku dimeru, který je žlutou kapalinou.[2][3][4]
| Rhodocen | |
|---|---|
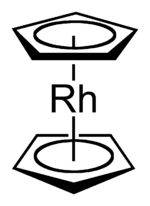
Strukturní vzorec | |
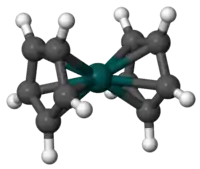
Model molekuly | |
| Obecné | |
| Systematický název | rhodocen |
| Sumární vzorec | C10H10Rh |
| Vzhled | žlutá pevná látka |
| Identifikace | |
| Registrační číslo CAS | 16941-11-0 |
| PubChem | 3082022 |
| SMILES | [cH-]1cccc1.[cH-]1cccc1.[Rh+2] |
| InChI | 1S/2C5H5.Rh/c2*1-2-4-5-3-1;/h2*1-5H;/q2*-1;+2 |
| Vlastnosti | |
| Molární hmotnost | 233,09 g/mol |
| Teplota tání | 174 °C (447 K) |
| Rozpustnost v polárních rozpouštědlech |
rozpustný v acetonitrilu |
| Rozpustnost v nepolárních rozpouštědlech |
mírně rozpustný v dichlormethanu |
| Není-li uvedeno jinak, jsou použity jednotky SI a STP (25 °C, 100 kPa). | |
| Některá data mohou pocházet z datové položky. | |
Do historie organokovové chemie patří objev Zeiseho soli[5][6][7] a tetrakarbonylu niklu v 19. století.[1] Tyto sloučeniny vzbudily zájem chemiků, protože neodpovídaly dosavadním modelům chemické vazby. Další výzvy se objevily s přípravou ferrocenu,[8] železnatého analogu rhodocenu a prvního známého metalocenu.[9] Ferrocen se ukázal jako neobvykle stabilní,[10] stejně jako jiné podobné struktury, například rhodocenium, kation odvozený od rhodocenu, a odpovídající struktury obsahující kobalt a iridium.[11] Studium organokovových sloučenin jako jsou výše uvedené vedlo k rozvoji nových modelů chemické vazby, které vysvětlovaly jejich tvorbu a stabilitu.[12][13] Díky zkoumání sendvičových sloučenin, jako je systém rhodocenium-rhodocen, získali Geoffrey Wilkinson a Ernst Otto Fischer v roce 1973 Nobelovu cenu za chemii.[14][15]
Díky své stabilitě a poměrně snadné přípravě jsou soli rhodocenia vhodným výchozím materiálem na přípravu rhodocenu a jeho substituovaných derivátů, které jsou nestabilní. Při původní syntéze byly použity cyklopentadienylový anion a tris(acetylacetonato)rhodium(III);[11] později bylo popsáno několik dalších postupů, například redox transmetalace v plynné fázi[16] a příprava s využitím polosendvičových prekurzorů.[17] Prvním substituovaným rhodocenem izolovaným za pokojové teploty, i když na vzduchu se rychle rozpadajícím, byl oktafenylrhodocen (derivát s osmi navázanými fenylovými skupinami). Pomocí rentgenové krystalografie bylo potvrzeno, že oktafenylrhodocen má sendvičovou strukturu v nezákrytové konformaci.[18] Na rozdíl od kobaltocenu, používaného ve výzkumu jako jednoelektronové redukční činidlo,[19] není žádný dosud známý derivát rhodocenu pro tyto účely dostatečně stabilní.
Deriváty rhodocenu lze použít na přípravu propojených metalocenů a následně zkoumat interakce mezi kovy;[20] tyto deriváty mohou mít využití například v molekulární elektronice a výzkumu mechanismů katalýzy.[21]
Historie

Objevy v organokovové chemii vedly k novým teoriím chemické vazby. Roku 1831 byla popsána Zeiseho sůl, K[PtCl3(C2H4)]·H2O[5] a v roce 1888 objevil Ludwig Mond tetrakarbonyl niklu.[24] V každé z těchto sloučenin se nacházela vazba mezi kovem a malou molekulou, kterou byl ethen u Zeiseho soli a oxid uhelnatý u tetrakarbonylu niklu.[6] Kalotový model aniontu Zeiseho soli (obrázek vlevo)[22][23] zobrazuje přímou vazbu platinového centra (znázorněného modře) a atomy uhlíku (černě) ethenového ligandu; podobné vazby kov-uhlík jsou typické pro organokovové sloučeniny. Modely chemických vazeb nedostačovaly k vysvětlení vlastností vazeb kov-alken, a to až do vytvoření Dewarova–Chattova–Duncansonova modelu v 50. letech 20. století.[12][7][25][26] Původní formulace se zaměřovala pouze na vazby kov-alken,[24] model byl však postupně rozšířen na systémy jako jsou karbonyly kovů (například [Ni(CO)4]), u kterých se vyskytuje π retrodonace.[26]
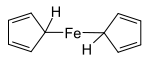

Ferrocen, [Fe(C5H5)2], byl poprvé připraven roku 1951 během pokusu o přípravu fulvalenu (C10H8) oxidativní dimerizací cyklopentadienu; vzniklý produkt měl sumární vzorec C10H10Fe a vyznačoval se „značnou stabilitou“.[10] Tento objev spustil zvýšený zájem o organokovovou chemii,[8][9] částečně proto, že struktura navržená Pausonem a Kealym neodpovídala tehdejším modelům chemické vazby a nedokázala vysvětlit neobvyklou stabilitu sloučeniny. Následně probíhaly pokusy o určení skutečné struktury ferrocenu, které by bylo možné popsat touto teorií. Sendvičovou strukturu popsaly tři skupiny: Robert Burns Woodward a Geoffrey Wilkinson zkoumali reaktivitu[27] a ukázali, že ferrocen vstupuje do podobných reakcí jako aromatické molekuly (například benzen),[28] Ernst Otto Fischer odvodil sendvičovou strukturu a připravil i několik dalších metalocenů, například kobaltocen,[29] a Eiland a Pepinsky potvrdil sendvičovou strukturu pomocí rentgenové krystalografie.[30] Aplikace teorie valenčních vazeb na ferrocen s uvažováním Fe2+ centra a dvou cyklopentadienidových aniontů (C5H5−), u kterých bylo známo, že jsou díky Hückelovu pravidlu aromatické a značně stabilní, umožnila správně předpovědět geometrii molekuly. S využitím teorie molekulových orbitalů se podařilo lépe zdůvodnit vysokou stabilitu ferrocenu.[13]
Vlastnosti kobaltocenu popsané Wilkinsonem a Fischerem ukázaly, že kobalticiniový kation [Co(C5H5)2]+ má podobnou stabilitu jako ferrocen, což se dalo očekávat vzhledem k tomu, že kobalticiniový kation je s ferrocenem isoelektronický. Na základě těchto pozorování se Wilkinson a F. Albert Cotton pokusili připravit soli rhodocenium and iridocenia.[11] Popsali syntézu řady rhodoceniových solí, jako jsou tribromid ([Rh(C5H5)2]Br3), chloristan ([Rh(C5H5)2]ClO4) a reineckát ([Rh(C5H5)2] [Cr(NCS)4(NH3)2]·H2O), a zjistili, že přidáním dipikrylaminu vzniká produkt o složení [Rh(C5H5)2] [N(C6H2N3O6)2].[11] V každém z těchto případů byl rhodoceniový kation velmi stabilní. Wilkinson and Fischer získali v roce 1973 společně Nobelovu cenu za chemii „za práci na chemii organokovových sloučenin“.[14][15]
Stabilitu metalocenů lze porovnávat pomocí redoxních potenciálů jednoelektronové redukce příslušného unipozitivního kationtu. Následující data byla získána pomocí nasycené kalomelové elektrody v acetonitrilu:
- [Fe(C5H5)2]+ / [Fe(C5H5)2] +0.38 V[31]
- [Co(C5H5)2]+ / [Co(C5H5)2] −0.94 V[2]
- [Rh(C5H5)2]+ / [Rh(C5H5)2] −1.41 V[2]
Tyto údaje poukazují na stabilitu neutrálního ferrocenu a kobaltoceniových a rhodoceniových kationtů. Rhodocen je přibližně o 500 mV lepším redukčním činidlem než kobaltocen, takže se snadněji oxiduje a je tak méně stabilní.[2] Při polarografickém zkoumání chloristanu rhodocenia za neutrálního pH se objevila katodová vlna s vrcholem na −1,53 V (oproti kalomelové elektrodě) na rtuťové kapkové elektrodě, což odpovídalo tvorbě rhodocenu v roztoku, avšak neutrální produkt se nepodařilo izolovat. Ve stejné studii se nepodařilo získat čistý iridocen z iridoceniových solí vystavených oxidačním podmínkám ani při zvýšeném pH.[11] Tato data naznačují, že rhodocen je vysoce nestabilní a iridocen by měl být ještě nestabilnější.
Příprava
Rhodoceniové soli byly poprvé popsány[11] dva roky po objevu ferrocenu.[10] Připraveny byly reakcemi karboaniontu Grignardova činidla cyklopentadienylmagnesiumbromidu (C5H5MgBr) s tris(acetylacetonátem) rhoditým (Rh(acac)3). Později byly vytvořeny rhodoceniové kationty redoxními transmetalačními reakcemi rhodných iontů s ferrocenem či niklocenem.[16]
- Rh+ + [(η5-C5H5)2M] → M + [(η5-C5H5)2Rh]+ M = Ni nebo Fe
Byly také popsány metody využívající mikrovlnné záření.[32] Reakcí cyklopentadienu s hydrátem chloridu rhoditého v methanolu a následným promytím methanolovým roztokem hexafluorofosforečnanu amonného se podařilo získat hexafluorofosforečnan rhodocenia; při reakci byla dosažena výtěžnost přes 60 % při vystavení reakční směsi mikrovlnám po dobu 30 sekund.[33]

Poté byl připraven redukcí rhodoceniových solí kapalným sodíkem i čistý rhodocen.[3] Když se do taveniny obsahující rhodoceniové ionty přidá sodík nebo draslík a provede se sublimace na povrch chlazený kapalným dusíkem, tak vzniká černá polykrystalické hmota.[34] Jejím zahříváním na pokojovou teplotu se tvoří žlutá pevná látka, která je dimerem rhodocenu. Podobným způsobem lze připravit také dimer iridocenu.[34]
Vlastnosti
Pravidlo 18 elektronů je ekvivalentem oktetového pravidla u chemických prvků; pomocí něj lze předpovídat stabilitu organokovových sloučenin.[35] Podle něj jsou organokovové částice "u kterých je součt poučtu valenčních elektronů kovu a elektronů dodávaných ligandy 18 are obvykle stabilní."[35] Tím lze vysvětlit neobvyklou stabilitu ferrocenu[10] a kobalticiniových a rhodoceniových kationtů[29] – mají podobné geometrie a jsou to isoelektronické struktury s 18 valenčními elektrony. Nestabilita rhodocenu a kobaltocenu vyplývá z toho, že jde o částice s 19 valenčními elektrony; tím se vysvětlují potíže při izolování rhodocenu z roztoků rhodoceniových solí.[11] Chemické vlastnosti rhodocenu jsou řízeny tendencemi získat 18elektronovou konfiguraci.
Rhodocen se jako [Rh(C5H5)2], paramagnetický 19elektronový monomerní radikál vyskytuje pouze při teplotách do −196 °C nebo nad 150 °C v plynném skupenství.[2][3][4] Tento monomer má typickou sendvičovou strukturu. Při 25 °C je poločas monomerní formy v acetonitrilu kratší než dvě sekundy;[2] a rhodocen vytváří [Rh(C5H5)2]2, a diamagnetický 18elektronový můstkový dimer s ansa-metalocenovou strukturou.[34] Pomocí elektronové paramagnetické rezonance (ESR), nukleární magnetické rezonance (NMR) a infračervené spektroskopie lze zkoumat rovnováhu mezi monomerní a dimerní formou.[4] ESR zobrazuje osu symetrie vysokého řádu (Cn, n > 2) s na ní kolmou zrcadlovou rovinou (σ) jako prvky symetrie; což naznačuje, že monomer má sendvičovou strukturu odpovídající metalocenu[3], i když interpretace těchto dat je sporná.[34] Rozklad monomeru byl také zkoumán hmotnostní spektrometrií.[36] Dimerizace je redoxní proces; v dimeru je rhodium v oxidačním čísle +I a v monomeru má oxidační číslo +II. Rhodium má ve stabilních sloučeninách obvykle oxidační číslo +I nebo +III[37]
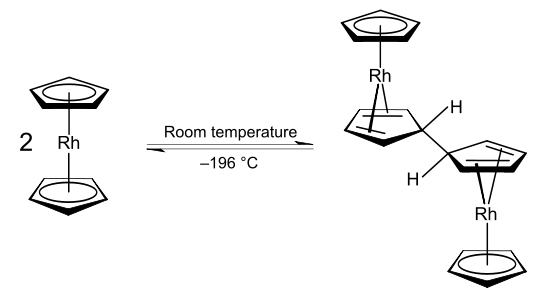
Tato dimerizace snižuje počet elektronů okolo centrálního atomu rhodia z 19 na 18, protože oxidativním párováním dvou cyklopentadienylových ligandů vzniká nový ligand s nižší hapticitou, který dodává na kovové centrum méně elektronů. Hapticita označuje počet uhlíkových (nebo jiných) atomů, na které se ligand (zde kovové centrum) váže (n)[38] a označuje se symbolem ηn; například ethenový ligand v Zeiseho soli je na platinové centrum navázán oběma atomy uhlíku a formálně tak má vzorec K[PtCl3(η2-C2H4)]·H2O.[6] Karbonylový ligand v tetrakarbonylu niklu se váže pouze přes jeden atom uhlíku a je tak monohapto ligandem. Cyklopentadienylové ligandy u mnoha metalocenů a polosendvičových sloučenin jsou pentahapto ligandy, proto se pro monomer rhodocenu používá vzorec [Rh(η5-C5H5)2]. Dimer, jenž je tvořen dvojicí spojených cyklopentadienylových ligandů, je čtyřelektronovým tetrahaptodonorem na každém rhodném centru. Vyšší stabilita 18elektronového rhodného dimeru oproti 19elektronovému rhodnatému monomeru je pravděpodobnou příčinou toho, že monomer lze zachytit pouze za extrémních podmínek.[2][4]
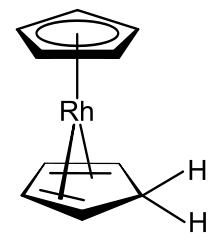
Cotton a Wilkinson zjistili,[11] že 18elektronový rhodoceniový kation [Rh(η5-C5H5)2]+, obsahující trojmocné rhodium, může být ve vodných roztocích redukován na monome; neutrální produkt nebylo možné izolovat nejen kvůli jeho dimerizaci, ale i kvůli tomu, že rhodnatý radikálový monomer může spontánně vytvářet stabilní rhodné sloučeniny se smíšenou hapticitou [(η5-C5H5)Rh(η4-C5H6)].[3] Rozdíly ve vlastnostech rhodocenu a tohoto derivátu spočívají v dvou záležitostech: (1) jeden z cyklopentadienylových ligandů formálně získává atom vodíku a stává se cyklopentadienem, který je stále navázán na kovové centrum, ovšem nově jako 4elektronový η4- donor. (2) Rhodnaté centrum se redukuje na rhodné. Tyto dvě změny způsobují, že derivát má 18 valenčních elektronů. Fischer uvažoval o tom, že k tvorbě iontu z tohoto derivátu dochází v oddělených protonačních a redukčních krocích, ovšem neposkytl pro tuto domněnku žádný důkaz.[3] Vzniklé (η4-Cyklopentadien)(η5-cyklopentadienyl)rhodium, je neobvyklým organokovovým komplexem obsahujícím jak cyklopentadienylový anion, tak i cyklopentadien jako ligandy. Zjistilo se, že tuto sloučeninu je možné připravit také redukcí roztoku rhodocenia ve vodném roztoku ethanolu pomocí tetrahydridoboritanu sodného; produkt byl charakterizován jako hydrid biscyklopentadienylrhodný.[39]
Fischer se svými spolupracovníky zkoumal rovněž chemické vlastnosti iridocenu, což je třetí člen řady obsahující rhodocen a kobaltocen, a zjistil, že se podobá rhodocenu. Popsal přípravu řady solí iridocenia, například tribromidu a hexafluorofosforečnanu.[4] Podobně jako rhodocen i iridocen za pokojové teploty tvoří dimer, ovšem monomer lze detekovat při nízkých teplotách a měření pomocí infračervené spektroskopie, spektroskopie nukleární magnetické rezonance a elektronové paramagnetické rezonance naznačují, že zde existuje chemická rovnováha, a potvrdily sendvičovou strukturu monomeru iridocenu.[3][4] Komplex [(η5-C5H5)Ir(η4-C5H6)], analog rhodocenového derivátu popsaného Fischerem,[3] má obdobné vlastnosti, pouze ovlivněné větší mírou π-retrodonace u iridných systémů oproti kobaltným a rhodným.[40]
Substituované rhodoceny a rhodoceniové ionty
Kation [(η5-C5tBu3H2)Rh(η5-C5H5)]+
Byly vyvinuty nové postupy přípravy substituovaných cyklopentadienylových komplexů využívající substituované vinylcyklopropeny.[41][42][43] Patří sem například zvětšování cyklů vinylcyklopropanovými přesmyky za vzniku cyklopentenů[44]. Kation [(η5-C5tBu3H2)Rh(η5-C5H5)]+ se připravuje posloupností reakcí, která začíná adicí dimeru chlorbisethylenrhodia, [(η2-C2H4)2Rh(μ-Cl)]2, na 1,2,3-tri-terc-butyl-3-vinyl-1-cyklopropen, po které proběhne reakce s cyklopentadienylthaliem:[41][42]
Rh(C5H5))BF4.svg.png.webp)
Rhodité pentadiendiylové komplexy s 18 valenčními elektrony vznikající při této reakci potvrzují nestabilitu rhodocenu, protože mohou být vstřikovány do toluenu několik měsíců, aniž by vznikal 1,2,3-tri-terc-butylrhodocen, ale za oxidujících podmínek se 1,2,3-tri-terc-butylrhodoceniový kation tvoří rychle.[41] K podrobnému prozkoumání tohoto (i jiných podobných) procesu byla použita cyklická voltametrie.[41][42] Mechanismus reakce se skládá z odštěpení jednoho elektronu z pentadiendiylového ligandu a následného rychlého přesmyku (při němž se odpojuje atom vodíku) za vzniku 1,2,3-tri-terc-butylrhodoceniového iontu.[42] Pomocí rentgenové krystalografie byla určena struktura tetrafluorboritanu i hexafluorofosforečnanu tohoto kationtu.[42]
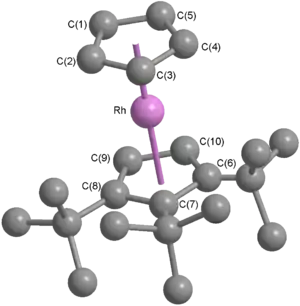
[(η5-C5tBu3H2)Rh(η5-C5H5)]BF4 vytváří bezbarvé centrosymetrické monoklinické krystaly s prostorovou grupou P21/c a hustotou 1,486 g/cm3.[42] Na ORTEP diagramu struktury tohoto iontu je vidět, že má geometrii, kterou lze u rhodocenu či rhodoceniového kationtu očekávat. Dva cyklopentadienylové kruhy jsou téměř rovnoběžné (úhel centroid–Rh–centroid je 177,2°) a kovové centrum je o něco blíže k substituovanému cyklopentadienylovému kruhu (vzdálenosti Rh–centroid jsou 1,819 Å a 1,795 Å), což se přičítá silnějšímu indukčnímu efektu terc-butylovaných ligandů.[42] ORTEP diagram také ukazuje, že kation má v pevném skupenství zákrytovou konformaci. Krystalová struktura hexafluorofosforečnanu zahrnuje tři krystalograficky nezávislé kationty, jeden v zákrytové, jeden v nezákrytové a jeden s narušenou rotací,[42] což naznačuje, že konformace je závislá na přítomných aniontech a také že energetická bariéra rotace je nízká – u ferrocenu jde o ~5 kJ mol−1 v roztoku i v plynném skupenství.[13]
Rh(C5H5))BF4.png.webp)
Výše uvedený diagram znázorňuje délky vazeb rhodium–uhlík (červeně, uvnitř pětiúhelníků vlevo) a uhlík–uhlík (modře, mimo pětiúhelníky vlevo) u obou ligandů, stejně jako vazebné úhly (zeleně, uvnitř pětiúhelníků vpravo) v každém cyklopentadienylovém kruhu. Atomy jsou zde označeny stejně jako u výše uvedené krystalové struktury. V nesubstituovaných cyklopentadienylových ligandech, se délky vazeb uhlík–uhlík pohybují od 1,35 Å do 1,40 Å a vnitřní úhly vazeb mezi 107° a 109°; u pravidelného pětiúhelníku se jedná o 108°. Vazby rhodium–uhlík mají délky od 2,16 Å do 2,18 Å.[42] Tyto hodnoty odpovídají η5-koordinaci ligandu na kovové centrum. U substitutovaného ligandu jsou rozdíly větší: vazby uhlík-uhlík mají délky mezi 1,39 Å a 1,48 Å, vnitřní úhly mezi 106° a 111°, a vazby rhodium–uhlík se vyznačují délkami mezi 2,14 Å a 2,20 Å. Větší rozdíly lze vysvětlit narušením struktury, které je nutné k omezení sterického napětí vytvářeného sousedními terc-butylovými substituenty; i přes tyto rozdíly je substituovaný cyklopentadienyl také η5-koordinovaný.[42]
Stabilitu metalocenů ovlivńují substituenty na cyklopentadienylových kruzích. Srovnáním redukčních potenciálů kobaltocenia a dekamethylkobaltocenia vychází, že dekamethylovaný derivát je o přibližně 600 mV silnějším redukčním činidlem než základní metalocen;[19] podobné výsledky se objevují i u ferrocenu[45] a rhodocenu.[46] Následující údaje se vztahují vzhledem k redoxnímu páru ferrocenium / ferrocen:[47]
| Poloreakce | E° (V) |
|---|---|
| [Fe(C5H5)2]+ + e− ⇌ [Fe(C5H5)2] | 0 (z definice) |
| [Fe(C5Me5)2]+ + e− ⇌ [Fe(C5Me5)2] | −0,59[45] |
| [Co(C5H5)2]+ + e− ⇌ [Co(C5H5)2] | −1,33[19] |
| [Co(C5Me5)2]+ + e− ⇌ [Co(C5Me5)2] | −1,94[19] |
| [Rh(C5H5)2]+ + e− ⇌ [Rh(C5H5)2] | −1,79[2] † |
| [Rh(C5Me5)2]+ + e− ⇌ [Rh(C5Me5)2] | −2,38[46] |
| [(C5tBu3H2)Rh(C5H5)]+ + e− ⇌ [(C5tBu3H2)Rh(C5H5)] | −1.83[42] |
| [(C5tBu3H2)Rh(C5Me5)]+ + e− ⇌ [(C5tBu3H2)Rh(C5Me5)] | −2,03 [42] |
| [(C5H5Ir(C5Me5)]+ + e− ⇌ [(C5H5Ir(C5Me5)] | −2,41[48] † |
| [Ir(C5Me5)2]+ + e− ⇌ [Ir(C5Me5)2] | −2,65[48] † |
Rozdíly potenciálů u kobaltoceniových částic jsou způsobeny indukčními efekty vyvolávanými alkylovými skupinami,[19] které dále stabilizují systémy s 18 valenčními elektrony. Podobný efekt se objevuje i u rhodocenia.[42] U substitutovaného iridocenia cyklická voltametrie naznačuje, že dochází k nevratné redukci i při teplotě okolo −60 °C;[48], zatímco redukce odpovídajících rhodocenů je při pokojové teplotě částečně a při −35 °C zcela vratná.[46] Nevratnost redukce substituvaného iridocenia je způsobována prudkou dimerizací vznikajícího produktu s 19 valenčními elektrony, což ukazuje na nižší stabilitu iridocenů ve srovnání s odpovídajícími rhodoceny.[48]
Pentasubstituované cyklopentadienylové ligandy
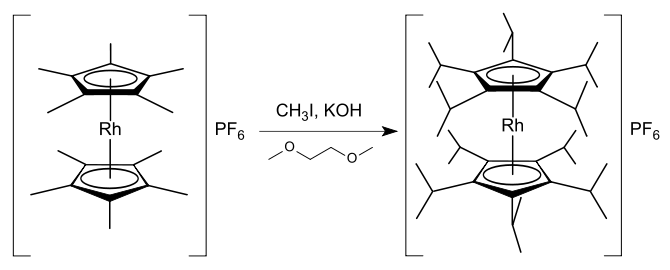
Jsou popsané pentasubstituované cyklopentadienylové sloučeniny, například organokovové komplexy pentamethylcyklopentadienylových a pentafenylcyklopentadienylových ligandů.[49] Substitucí cyklopentadienylových kruhů rhodocenu a rhodoceniových solí vznikají sloučeniny, které jsou díky zvýšené delokalizaci kladného náboje či elektronové hustoty stabilnější a vytvářejí sterické napětí vůči ostatním molekulám okolo kovového centra.[36] Je známo mnoho mono- a disubstituovaných sloučenin rhodocenia, ovšem bez výraznější substituce nelze dosáhnout významné stabilizace.[36] Ke známým vysoce substituovaným rhodoceniovým solím patří například hexafluorofosforečnany dekamethylrhodocenia [(η5-C5Me5)2Rh]PF6,[50] dekaisopropylrhodocenia [(η5-C5iPr5)2Rh]PF6,[51] a oktafenylrhodocenia [(η5-C5Ph4H)2Rh]PF6.[18] Tetrafluoroboritan dekamethylrhodocenia se dá připravit z komplexu tris(acetonu) [(η5-C5Me5)Rh(Me2CO)3](BF4)2 reakcí s pentamethylcyklopentadienem; jsou známy i obdobné metody přípravy příslušných komplexů iridia.[52] Hexafluorofosforečnan dekaisopropylrhodicinia je možné získat reakcí v 1,2-dimethoxyethanu za tvorby 20 vazeb uhlík–uhlík:[51]
Podobnou reakcí lze připravit hexafluorofosforečnan pentaisopropylrhodocenia [(η5-C5iPr5)Rh(η5-C5H5)]PF6 z hexafluorofosforečnanu pentamethylrhodocenia [(η5-C5Me5)Rh(η5-C5H5)]PF6 s 80% výtěžností.[51] Tyto reakce dokazují, že kyselost methylových vodíků v pentamethylcyklopentadienylových komplexech lze výrazně zvýšit pomocí kovového centra. Mechanisticky reakce probíhá tak, že hydroxid draselný deprotonuje methylovou skupinu a vzniklý karboanion je nukleofilně atakován jodmethanem za tvorby nové vazby uhlík–uhlík.[51]
Byly také popsány tetrafluoroboritan pentafenylrhodocenia [(η5-C5Ph5)Rh(η5-C5H5)]BF4 a pentamethylpentafenylrhodocenia [(η5-C5Ph5)Rh(η5-C5Me5)]BF4. Tyto sloučeniny ukazují, že sendvičové sloučeniny rhodia lze připravit z polosendvičových prekurzorů; například postupem podobným tris(aceton)ové syntéze tetrafluoroboritanu dekamethylrhodocenia,[52] byl získán tetrafluoroboritan pentafenylrhodocenia ze soli tris(acetonitril)u [(η5-C5Ph5)Rh(CH3CN)3](BF4)2 reakcí s cyklopentadienidem sodným:[17]
- [(η5-C5Ph5)Rh(MeCN)3](BF4)2 + NaC5H5 → [(η5-C5Ph5)Rh(η5-C5H5)]BF4 + NaBF4 + 3 MeCN

Prvním popsaným derivátem rhodocenu izolovaným za pokojové teploty je oktafenylrhodocen, [(η5-C5Ph4H)2Rh]. Jedná se o látku tvořící olivově zelené krystaly, které se v roztoku rychle rozkládají, na vzduchu v několika minutách, což naznačuje výrazně vyšší citlivost na vzduch než u odpovídajícího komplexu kobaltu, i když je látka mnohem stabilnější než samotný rhodocen. Rozdíl je způsoben nižší stabilitou oxidačního čísla II u rhodia oproti kobaltu.[18][37] Redukční potenciál kationtu [(η5-C5Ph4H)2Rh]+ (v dimethylformamidu, vůči páru ferrocenium/ferrocen) je −1,44 V, což je v souladu s větší termodynamickou stabilizací rhodocenu ligandy C5HPh4 vůči C5H5 nebo C5Me5 ligandům.[18] Kobaltocen je dobrým jednoelektronovým redukčním činidlem pro laboratorní využití, díky své rozpustnosti v nepolárních organických rozpouštědlech[19] a jeho redoxní pár má vhodné vlastnosti k tomu, aby mohl být použit jako interní standard v cyklické voltametrii.[53] Žádný dosud připravený substituovaný rhodocen není k tomuto použití dostatečně stabilní.
Syntéza oktafenylrhodocenu probíhá ve třech krocích: refluxu diglymu, přidání kyseliny hexafluorofosforečné a redukci sodným amalgámem v tetrahydrofuranu:[18]
- Rh(acac)3 + 2 KC5Ph4H → [(η5-C5Ph4H)2Rh]+ + 2 K+ + 3 acac−
- [(η5-C5Ph4H)2Rh]+ + 3 acac− + 3 HPF6 → [(η5-C5Ph4H)2Rh]PF6 + 3 Hacac + 2 PF6−
- [(η5-C5Ph4H)2Rh]PF6 + Na/Hg → [(η5-C5Ph4H)2Rh] + NaPF6
Krystalová struktura oktafenylrhodocenu se vyznačuje nezákrytovou konformací[18] (podobně jako u ferrocenu).[13] Vzdálnost rhodium–centroid činí 190,4 pm a průměrná délka vazeb rhodium–uhlík je 226 pm; vazby uhlík–uhlík jsou v průměru 144 pm dlouhé.[18] Tyto vzdálenosti jsou podobné jako u 1,2,3-tri-terc-butylrhodoceniového kationtu, s tím, že efektivní velikost rhodiového centra je větší v důsledku většího iontového poloměru u Rh2+ než u Rh3+.
Použití
Biomedicínské využití derivátů
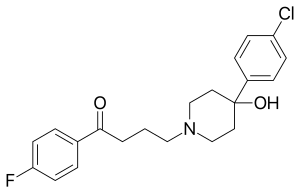
Metalofarmaka, léčiva patřící mezi organokovové sloučeniny, jsou předmětem mnoha studií,[54][55] zkoumá se i lékařské využití sloučenin rhodia.[56] Velká část výzkumu byla zaměřena na využití léčiv obsahujících deriváty metalocenů ruthenia[57] a železa[58]. Příkladem je použití metalocenů namísto fluorofenylové skupiny v molekule haloperidolu,[59], používaného jako antipsychotikum. Zkoumaný ferrocenylový derivát haloperidolu má chemický vzorec (C5H5)Fe(C5H4)–C(=O)–(CH2)3–N(CH2CH2)2C(OH)–C6H4Cl a může být přeměněn na ruthenatý analog pomocí transmetalační reakce. Pomocí radioizotopu 103Ru lze vytvořit ruthenocenylhaloperidolové radiofarmakum, které mají u myší a krys vysokou afinitu k plicním a nízkou afinitou k mozkovým tkáním.[59] Beta přeměnou 103Ru vzniká v rhodocenyl–haloperidolu metastabilní nuklid 103mRh. Tato sloučenina, podobně jako ostatní deriváty rhodocenu, má nestabilní elektronovou konfiguraci s 19 valečními elektrony a rychle se oxiduje na kationtový rhodocenium–haloperidolový produkt.[59] Předmětem studií byla separace ruthenocenyl–haloperidolu a rhodocenium–haloperidolu a jejich distribuce v jednotlivých orgánech.[60] 103mRh má poločas přeměny 56 min a vyzařuje záření gama o energii 39,8 keV a krátce po beta přeměně izotopu ruthenia tak následuje emise gama fotonů z izotopu rhodia. Dalšími beta- a gama radionuklidy s lékařským využitím patří 131I, 59Fe, 47Ca a 103mRh mají možné využití v radioterapii[56]
Interakce kov–kov u propojených metalocenů
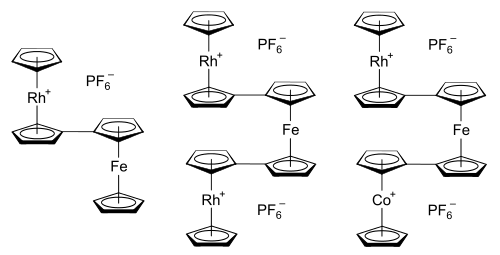
Původní motivací k výzkumu rhodocenových systémů byla snaha porozumět povaze a vazbám u metalocenů. Později se hlavní zájem přesunul k cíli prozkoumat a najít využití pro interakce mezi kovy u propojených metalocenů.[20] Možné využití takových systémů zahrnuje například molekulární elektroniku,[21] polovodivé (a možná i ferromagnetické) polymery metallocenů,[20] a průzkum hranic mezi heterogenní a homogenní katalýzou.[21] Jako příklady bimetalocenů a termetalocenů obsahujících rhodocenylové skupiny lze uvést hexafluorofosforečnany rhodocenylferrocenu, 1,1'-dirhodocenylferrocenu a 1-kobaltocenyl-1'-rhodocenylferrocenu.[61] Propojené metaloceny lze připravit napojováním několika metalocenylových skupin na jeden cyklopentadienylový ligand.[21]
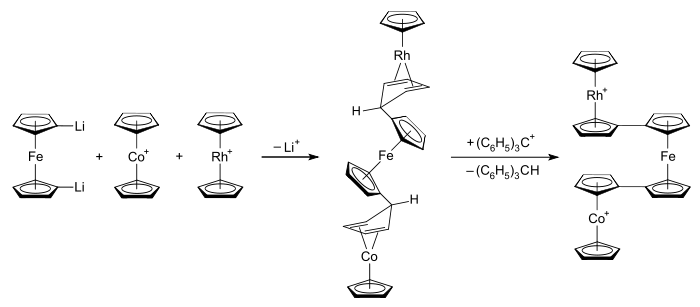
Strukturní studie termetalocenových systémů ukázaly, že tyto sloučeniny obvykle mají „zákrytovou dvojitě transoidovou“ geometrii.[62] Příkladem může být 1-kobaltocenyl-1'-rhodocenylferrocenový kation (znázorněný výše), ve kterém jsou kobaltocenylová a rhodocenylová skupina navzájem v zákrytové konformaci a uhlíkové atomy v pozicích 1 a 1' na ferrocenovém jádru jsou vertikálně uspořádané tak blízko k sobě, jak je možné a zaujímají k cyklopentadienylovým cyklům v metalocenových jednotkách nezákrytové konformace.[62] Při přípravě těchto termetalocenů se smíchají roztoky rhodoceniových a kobaltoceniových solí s 1,1'-dilithioferrocenem. Tímto se utvoří nenabitý meziprodukt s propojenými cyklopentadienyl–cyklopentadienovými ligandy navázanými na sebe podobně jako u dimeru rhodocenu. Tyto ligandy následně reagují s trifenylmethylovými karbokationty za vzniku soli termetalocenu, [(η5-C5H5)Rh(μ-η5:η5-C5H4–C5H4)Fe(μ-η5:η5-C5H4–C5H4)Co(η5-C5H5)](PF6)2. Postup je zobrazen na obrázku níže:[61][62]
Polymeru obsahující rhodocenium
První polymery obsahující rhodocenium v postranních řetězcích byly připraveny kontrolovanými polymerizačními metodami, jako jsou vratná adičně−fragmentační polymerizace s přesunem řetězce (RAFT) a metatezní polymerizace s otevíráním kruhu (ROMP).[63]
Reference
V tomto článku byl použit překlad textu z článku Rhodocene na anglické Wikipedii.
- CRABTREE, R. H. The Organometallic Chemistry of the Transition Metals. 5th. vyd. Hoboken, NJ: John Wiley and Sons, 2009. Dostupné online. ISBN 978-0-470-25762-3. S. 2. (anglicky)
- EL MURR, N.; SHEATS, J. E.; GEIGER, W. E.; HOLLOWAY, J. D. L. Electrochemical Reduction Pathways of the Rhodocenium Ion. Dimerization and Reduction of Rhodocene. Inorganic Chemistry. 1979, s. 1443–1446. DOI 10.1021/ic50196a007. (anglicky)
- FISCHER, E. O.; WAWERSIK, H. Über Aromatenkomplexe von Metallen. LXXXVIII. Über Monomeres und Dimeres Dicyclopentadienylrhodium und Dicyclopentadienyliridium und Über Ein Neues Verfahren Zur Darstellung Ungeladener Metall-Aromaten-Komplexe. Journal of Organometallic Chemistry. 1966, s. 559–567. DOI 10.1016/S0022-328X(00)85160-8. (německy)
- KELLER, H. J.; WAWERSIK, H. Spektroskopische Untersuchungen an Komplexverbindungen. VI. EPR-spektren von (C5H5)2Rh und (C5H5)2Ir. Journal of Organometallic Chemistry. 1967, s. 185–188. DOI 10.1016/S0022-328X(00)84718-X. (německy)
- ZEISE, W. C. Von der Wirkung zwischen Platinchlorid und Alkohol, und von den dabei entstehenden neuen Substanzen. Annalen der Physik. 1831, s. 497–541. Dostupné online. DOI 10.1002/andp.18310970402. Bibcode 1831AnP....97..497Z. (německy)
- HUNT, L. B. The First Organometallic Compounds: William Christopher Zeise and his Platinum Complexes. Platinum Metals Review. 1984, s. 76–83. Dostupné online. (anglicky)
- WINTERTON, N. Modern Coordination Chemistry: The Legacy of Joseph Chatt. Redakce Leigh G. J.. [s.l.]: RSC Publishing, 2002. ISBN 9780854044696. Kapitola Some Notes on the Early Development of Models of Bonding in Olefin-Metal Complexes, s. 103–110. (anglicky)
- LASZLO, P.; HOFFMANN, R. Ferrocene: Ironclad History or Rashomon Tale?. Angewandte Chemie International Edition. 2000, s. 123–124. DOI 10.1002/(SICI)1521-3773(20000103)39:1<123::AID-ANIE123>3.0.CO;2-Z. PMID 10649350. (anglicky)
- FEDERMAN NETO, A.; PELEGRINO, A. C.; DARIN, V. A. Ferrocene: 50 Years of Transition Metal Organometallic Chemistry — From Organic and Inorganic to Supramolecular Chemistry. ChemInform. 2004. DOI 10.1002/chin.200443242. (anglicky) (Abstract; original published in Trends in Organometallic Chemistry, 4:147–169, 2002)
- KEALY, T. J.; PAUSON, P. L. A New Type of Organo-Iron Compound. Nature. 1951, s. 1039–1040. DOI 10.1038/1681039b0. S2CID 4181383. Bibcode 1951Natur.168.1039K. (anglicky)
- COTTON, F. A.; WHIPPLE, R. O.; WILKINSON, G. Bis-Cyclopentadienyl Compounds of Rhodium(III) and Iridium(III). Journal of the American Chemical Society. 1953, s. 3586–3587. DOI 10.1021/ja01110a504. (anglicky)
- MINGOS, D. M. P. A Historical Perspective on Dewar's Landmark Contribution to Organometallic Chemistry. Journal of Organometallic Chemistry. 2001, s. 1–8. DOI 10.1016/S0022-328X(01)01155-X. (anglicky)
- Mehrotra, R. C.; SINGH, A. Organometallic Chemistry: A Unified Approach. 2nd. vyd. New Delhi: New Age International, 2007. Dostupné online. ISBN 978-81-224-1258-1. S. 261–267. (anglicky)
- The Nobel Prize in Chemistry 1973 [online]. Nobel Foundation [cit. 2010-09-12]. Dostupné online. (anglicky)
- SHERWOOD, Martin. Metal Sandwiches. New Scientist. 1 November 1973, s. 335. Dostupné online [cit. 17 June 2017]. (anglicky)
- JACOBSON, D. B.; BYRD, G. D.; FREISER, B. S. Generation of Titanocene and Rhodocene Cations in the Gas Phase by a Novel Metal-Switching Reaction. Journal of the American Chemical Society. 1982, s. 2320–2321. DOI 10.1021/ja00372a041. (anglicky)
- HE, H. T. Synthesis and Characterisation of Metallocenes Containing Bulky Cyclopentadienyl Ligands. University of Sydney: [s.n.], 1999. OCLC 222646266 (anglicky)
- COLLINS, J. E.; CASTELLANI, M. P.; RHEINGOLD, A. L.; MILLER, E. J.; GEIGER, W. E.; RIEGER, A. L.; RIEGER, P. H. Synthesis, Characterization, and Molecular-Structure of Bis(tetraphenylcyclopentadienyl)rhodium(II). Organometallics. 1995, s. 1232–1238. DOI 10.1021/om00003a025. (anglicky)
- CONNELLY, N. G.; GEIGER, W. E. Chemical Redox Agents for Organometallic Chemistry. Chemical Reviews. 1996, s. 877–910. DOI 10.1021/cr940053x. PMID 11848774. (anglicky)
- BARLOW, S.; O'HARE, D. Metal–Metal Interactions in Linked Metallocenes. Chemical Reviews. 1997, s. 637–670. DOI 10.1021/cr960083v. PMID 11848884. (anglicky)
- WAGNER, M. A New Dimension in Multinuclear Metallocene Complexes. Angewandte Chemie International Edition. 2006, s. 5916–5918. DOI 10.1002/anie.200601787. PMID 16906602. (anglicky)
- BLACK, M.; MAIS, R. H. B.; OWSTON, P. G. The crystal and molecular structure of Zeise's salt, KPtCl3.C2H4.H2O. Acta Crystallographica B. 1969, s. 1753–1759. DOI 10.1107/S0567740869004699. (anglicky)
- JARVIS, J. A. J.; KILBOURN, B. T.; OWSTON, P. G. A Re-determination of the Crystal and Molecular Structure of Zeise's salt, KPtCl3.C2H4.H2O. Acta Crystallographica B. 1971, s. 366–372. DOI 10.1107/S0567740871002231. (anglicky)
- Modern Coordination Chemistry: The Legacy of Joseph Chatt. Redakce Leigh G. J.. Cambridge, UK: RSC Publishing, 2002. ISBN 0-85404-469-8. Kapitola Section D: Transition Metal Complexes of Olefins, Acetylenes, Arenes and Related Isolobal COmpounds, s. 101–110. (anglicky)
- MINGOS, D. Michael P. A Historical Perspective on Dewar's Landmark Contribution to Organometallic Chemistry. Journal of Organometallic Chemistry. 2001, s. 1–8. DOI 10.1016/S0022-328X(01)01155-X. (anglicky)
- ASTRUC, D. Organometallic Chemistry and Catalysis. Berlin: Springer, 2007. Dostupné online. ISBN 978-3-540-46128-9. S. 41–43. (anglicky)
- WILKINSON, G.; ROSENBLUM, M.; WHITING, M. C.; WOODWARD, R. B. The Structure of Iron Bis-Cyclopentadienyl. Journal of the American Chemical Society. 1952, s. 2125–2126. DOI 10.1021/ja01128a527. (anglicky)
- WERNER, H. Landmarks in Organo-Transition Metal Chemistry: A Personal View. New York: Springer Science, 2008. Dostupné online. ISBN 978-0-387-09847-0. S. 161–163. (anglicky)
- FISCHER, E. O.; PFAB, W. Zur Kristallstruktur der Di-Cyclopentadienyl-Verbindungen des zweiwertigen Eisens, Kobalts und Nickels. Zeitschrift für anorganische und allgemeine Chemie. 1952, s. 377–379. DOI 10.1002/zaac.19532740603. (německy)
- EILAND, P. F.; PEPINSKY, R. X-ray Examination of Iron Biscyclopentadienyl. Journal of the American Chemical Society. 1952, s. 4971. DOI 10.1021/ja01139a527. (anglicky)
- PAVLISHCHUK, V. V.; ADDISON, A. W. Conversion Constants for Redox Potentials Measured Versus Different Reference Electrodes in Acetonitrile Solutions at 25 °C. Inorganica Chimica Acta. 2000, s. 97–102. DOI 10.1016/S0020-1693(99)00407-7. (anglicky)
- BAGHURST, D. R.; MINGOS, D. M. P. Design and Application of a Reflux Modification for the Synthesis of Organometallic Compounds Using Microwave Dielectric Loss Heating Effects. Journal of Organometallic Chemistry. 1990, s. C57–C60. DOI 10.1016/0022-328X(90)87135-Z. (anglicky)
- BAGHURST, D. R.; MINGOS, D. M. P.; WATSON, M. J. Application of Microwave Dielectric Loss Heating Effects for the Rapid and Convenient Synthesis of Organometallic Compounds. Journal of Organometallic Chemistry. 1989, s. C43–C45. DOI 10.1016/0022-328X(89)85418-X. (anglicky)
- DE BRUIN, B.; HETTERSCHEID, D. G. H.; KOEKKOEK, A. J. J.; GRÜTZMACHER, H. The Organometallic Chemistry of Rh–, Ir–, Pd–, and Pt–Based Radicals: Higher Valent Species. Progress in Inorganic Chemistry. 2007, s. 247–354. Dostupné online. ISBN 978-0-471-68242-4. DOI 10.1002/9780470144428.ch5. (anglicky)
- KOTZ, J. C.; TREICHEL, P. M.; TOWNSEND, J. R. Chemistry and Chemical Reactivity, Volume 2. 7th. vyd. Belmont, CA: Cengage Learning, 2009. Dostupné online. ISBN 978-0-495-38703-9. S. 1050–1053. (anglicky)
- ZAGOREVSKII, D. V.; HOLMES, J. L. Observation of Rhodocenium and Substituted-Rhodocenium Ions and their Neutral Counterparts by Mass Spectrometry. Organometallics. 1992, s. 3224–3227. DOI 10.1021/om00046a018. (anglicky)
- COTTON, S. A. Chemistry of Precious Metals. London: Blackie Academic and Professional, 1997. ISBN 0-7514-0413-6. Kapitola Rhodium and Iridium, s. 78–172. (anglicky)
- HILL, A. F. Organotransition Metal Chemistry. Cambridge, UK: Royal Society of Chemistry, 2002. Dostupné online. ISBN 0-85404-622-4. S. 4–7. (anglicky)
- GREEN, M. L. H.; PRATT, L.; WILKINSON, G. 760. A New Type of Transition Metal–Cyclopentadiene Compound. Journal of the Chemical Society. 1959, s. 3753–3767. DOI 10.1039/JR9590003753. (anglicky)
- SZAJEK, L. P.; SHAPLEY, J. R. Unexpected Synthesis of CpIr(η4-C5H6) and a Proton and Carbon-13 NMR Comparison with its Cobalt and Rhodium Congeners. Organometallics. 1991, s. 2512–2515. DOI 10.1021/om00053a066. (anglicky)
- DONOVAN-MERKERT, B. T.; TJIONG, H. I.; RHINEHART, L. M.; RUSSELL, R. A.; MALIK, J. Facile, Redox-Promoted Formation of Rhodocenium Complexes Bearing the 1,2,3-Tri-tert-butylcyclopentadienyl Ligan. Organometallics. 1997, s. 819–821. DOI 10.1021/om9608871. (anglicky)
- DONOVAN-MERKERT, B. T.; CLONTZ, C. R.; RHINEHART, L. M.; TJIONG, H. I.; CARLIN, C. M.; CUNDARI, Thomas R.; RHEINGOLD, Arnold L. Rhodocenium Complexes Bearing the 1,2,3-Tri-tert-butylcyclopentadienyl Ligand: Redox-Promoted Synthesis and Mechanistic, Structural and Computational Investigations. Organometallics. 1998, s. 1716–1724. DOI 10.1021/om9707735. (anglicky)
- Hughes, R. P.; TRUJILLO, H. A.; EGAN, J. W.; RHEINGOLD, A. L. Skeletal Rearrangement during Rhodium-Promoted Ring Opening of 1,2-Diphenyl-3-vinyl-1-cyclopropene. Preparation and Characterization of 1,2- and 2,3-Diphenyl-3,4-pentadienediyl Rhodium Complexes and Their Ring Closure to a 1,2-Diphenylcyclopentadienyl Complex. Organometallics. 1999, s. 2766–2772. DOI 10.1021/om990159o. (anglicky)
- GOLDSCHMIDT, Z.; CRAMMER, B. Vinylcyclopropane Rearrangements. Chemical Society Reviews. 1988, s. 229–267. DOI 10.1039/CS9881700229. (anglicky)
- NOVIANDRI, I.; BROWN, K. N.; FLEMING, D. S.; GULYAS, P. T.; LAY, P. A.; MASTERS, A. F.; PHILLIPS, L. The Decamethylferrocenium/Decamethylferrocene Redox Couple: A Superior Redox Standard to the Ferrocenium/Ferrocene Redox Couple for Studying Solvent Effects on the Thermodynamics of Electron Transfer. Journal of Physical Chemistry B. 1999, s. 6713–6722. DOI 10.1021/jp991381+. (anglicky)
- GUSEV, O. V.; DENISOVICH, L. I.; PETERLEITNER, M. G.; RUBEZHOV, A. Z.; USTYNYUK, Nikolai A.; MAITLIS, P. M. Electrochemical Generation of 19- and 20-electron Rhodocenium Complexes and Their Properties. Journal of Organometallic Chemistry. 1993, s. 219–222. DOI 10.1016/0022-328X(93)83193-Y. (anglicky)
- Gagne, R. R.; KOVAL, C. A.; LISENSKY, G. C. Ferrocene as an Internal Standard for Electrochemical Measurements. Inorganic Chemistry. 1980, s. 2854–2855. DOI 10.1021/ic50211a080. (anglicky)
- GUSEV, O. V.; PETERLEITNER, M. G.; IEVLEV, M. A.; KAL'SIN, A. M.; PETROVSKII, P. V.; DENISOVICH, L. I.; USTYNYUK, Nikolai A. Reduction of Iridocenium Salts [Ir(η5-C5Me5)(η5-L)]+ (L= C5H5, C5Me5, C9H7); Ligand-to-Ligand Dimerisation Induced by Electron Transfer. Journal of Organometallic Chemistry. 1997, s. 95–100. DOI 10.1016/S0022-328X(96)06675-2. (anglicky)
- OKUDA, J. Transition Metal Coordination Chemistry. Redakce W. A. Herrmann. Berlin: Springer-Verlag, 1992. (Topics in Current Chemistry; sv. 160). ISBN 3-540-54324-4. DOI 10.1007/3-540-54324-4_3. Kapitola Transition-Metal Complexes of Sterically Demanding Cyclopentadienyl Ligands, s. 97–145. (anglicky)
- KÖLLE, U.; KLÄUI, W. Z.l. Darstellung und Redoxverhalten einer Serie von Cp*/aqua/tripod-Komplexen des Co, Rh und Ru. Zeitschrift für Naturforschung B. 1991, s. 75–83. DOI 10.1515/znb-1991-0116. S2CID 95222717. (německy)
- BUCHHOLZ, D.; ASTRUC, D. The First Decaisopropylmetallocene – One-Pot Synthesis of [Rh(C5iPr5)2]PF6 from [Rh(C5Me5)2]PF6 by Formation of 20 Carbon–Carbon Bonds. Angewandte Chemie International Edition. 1994, s. 1637–1639. DOI 10.1002/anie.199416371. (anglicky)
- Gusev, O. V.; MOROZOVAA, L. N.; PEGANOVAA, T. A.; PETROVSKIIA, P. V.; USTYNYUKA N. A.; MAITLIS, P. M. Synthesis of η5-1,2,3,4,5-Pentamethylcyclopentadienyl-Platinum Complexes. Journal of Organometallic Chemistry. 1994, s. 359–363. DOI 10.1016/0022-328X(94)80223-8. (anglicky)
- STOJANOVIC, R. S.; BOND, A. M. Examination of Conditions under which the Reduction of the Cobaltocenium Cation can be used as a Standard Voltammetric Reference Process in Organic and Aqueous Solvents. Analytical Chemistry. 1993, s. 56–64. DOI 10.1021/ac00049a012. (anglicky)
- Clarke, M. J.; SADLER, P. J. Metallopharmaceuticals: Diagnosis and therapy. Berlin: Springer, 1999. ISBN 3-540-65308-2. (anglicky)
- JONES, C. J.; THORNBACK, J. Medicinal Applications of Coordination Chemistry. Cambridge, UK: RSC Publishing, 2007. ISBN 978-0-85404-596-9. (anglicky)
- PRUCHNIK, F. P. Metallotherapeutic Drugs and Metal-Based Diagnostic Agents: The Use of Metals in Medicine. Redakce Gielen M.. Hoboken, NJ: Wiley, 2005. ISBN 0-470-86403-6. DOI 10.1002/0470864052.ch20. Kapitola 45Rh — Rhodium in Medicine, s. 379–398. (anglicky)
- CLARKE, M. J. Ruthenium Metallopharmaceuticals. Coordination Chemistry Reviews. 2002, s. 69–93. DOI 10.1016/S0010-8545(02)00025-5. (anglicky)
- FOUDA, M. F. R.; ABD-ELZAHER, M. M.; ABDELSAMAIA, R. A.; LABIB, A. A. On the Medicinal Chemistry of Ferrocene. Applied Organometallic Chemistry. 2007, s. 613–625. DOI 10.1002/aoc.1202. (anglicky)
- WENZEL, M.; WU, Y. Ferrocen-, Ruthenocen-bzw. Rhodocen-analoga von Haloperidol Synthese und Organverteilung nach Markierung mit 103Ru-bzw. 103mRh. International Journal of Radiation Applications and Instrumentation A. 1988, s. 1237–1241. DOI 10.1016/0883-2889(88)90106-2. PMID 2851003. (německy)
- WENZEL, M.; WU, Y. F. Abtrennung von [103mRh]Rhodocen-Derivaten von den Analogen [103Ru]Ruthenocen-Derivaten und deren Organ-Verteilung. International Journal of Radiation Applications and Instrumentation A. 1987, s. 67–69. DOI 10.1016/0883-2889(87)90240-1. PMID 3030970. (německy)
- ANDRE, M.; SCHOTTENBERGER, H.; TESSADRI, R.; INGRAM, G.; JAITNER, P.; SCHWARZHANS, K. E. Synthesis and Preparative HPLC-Separation of Heteronuclear Oligometallocenes. Isolation of Cations of Rhodocenylferrocene, 1,1'-Dirhodocenylferrocene, and 1-Cobaltocenyl-1'-rhodocenylferrocene. Chromatographia. 1990, s. 543–545. DOI 10.1007/BF02269802. S2CID 93898229. (anglicky)
- JAITNER, P.; SCHOTTENBERGER, H.; GAMPER, S.; OBENDORF, D. Termetallocenes. Journal of Organometallic Chemistry. 1994, s. 113–120. DOI 10.1016/0022-328X(94)84013-X. (anglicky)
- YAN, Y.; DEATON, T. M.; ZHANG, J.; HONGKUN, H.; HAYAT, J.; PAGENI, P.; MATYJASZEWSKI, K. The Syntheses of Monosubstituted Rhodocenium Derivatives, Monomers and Polymers. Macromolecules. 2015, s. 1644–1650. DOI 10.1021/acs.macromol.5b00471. Bibcode 2015MaMol..48.1644Y. (anglicky)