Organické sloučeniny rhodia
Organické sloučeniny rhodia jsou organokovové sloučeniny obsahující vazby mezi atomy rhodia a uhlíku; hlavní využití mají jako katalyzátory organických reakcí.[1]
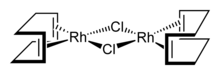
Stabilní organorhodiové sloučeniny i přechodné organorhodiové meziprodukty slouží jako katalyzátory hydroformylace, hydrogenace a izomerizace alkenů[2]
Rozdělení podle oxidačních čísel
Organokovové sloučeniny rhodia mají podobné vlastnosti jako odpovídající sloučeniny iridia, organokovovým sloučeninám kobaltu se podobají méně. Rhodium se může vyskytovat v oxidačních číslech od −III do +V, nejčastěší je RhI a RhIII. Rhodné sloučeniny (s konfigurací d8) obvykle mají rovinné čtvercové nebo trigonálně bipyramidové geometrie, rhodité (d6) jsou většinou oktaedrické.[2]
Rh0
Komplexy rhodia v oxidačním čísle 0 jsou binární karbonyly, jako příklad lze uvést dodekarbonyl tetrarhodia, Rh4(CO)10, a hexadekakarbonyl hexarhodia, Rh6(CO)16. Tyto sloučeniny lze získat redukční karbonylací rhoditých solí nebo Rh2Cl2(CO)4. Na rozdíl od obdobného Co2(CO)8 je Rh2(CO)8 velmi nestálý.
RhI
Rhodné komplexy jsou významnými homogenními katalyzátory. Takto se využívají například bis(trifenylfosfin)rhodiumkarbonylchlorid, dimery chlorobis(ethylen)rhodia, cyklooctadienrhodiumchloridu a chlorobis(cyklookten)rhodia, dikarbonyl(acetylacetonáto)rhodium(I) a rhodiumkarbonylchlorid. K významným katalyzátorům patří i Wilkinsonův katalyzátor (RhCl(PPh3)3), jenž podle definice nepatří mezi organokovové sloučeniny. Jednoduché alkenové komplexy, dimery chlorobis(ethylen)rhodia, chlorobis(cyklookten)rhodia a cyklooktadienrhodia, se často používají jako zdroje „RhCl“, přičemž se využívá nestálost alkenových ligandů a jejich snadné odstranění hydrogenačnímí reakcemi. (η5-Cp)RhL2 se připúravuje z Rh2Cl2L4 (L = CO, C2H4).
RhII
Na rozdíl od běžně se vyskytujících kobaltnatých komplexů jsou komplexy dvojmocného rhodia vzácné. Jako příklad sloučeniny lze uvést rhodocen, i když se vyskytuje v rovnováze s dimerem obsahujícím jednomocné rhodium. I když není organokovový, tak octan rhodnatý (Rh2(OAc)4) katalyzuje cyklopropanace přes organokovové meziprodukty. Rhodnaté porfyrinové komplexy reagují s methanem.[3]
RhIII
Rhodium bývá nejčastěji dodáváno v oxidačním čísle III, výchozím reaktantem je nejčastěji hydratovaný chlorid rhoditý, jenž reaguje s alkeny a oxidem uhelnatým za tvorby organokovových komplexů, přičemž často dochází k redukci na RhI. Existují také cyklopentadienylové komplexy rhodia, jako je polosendvičový dimer pentamethylcyklopentadienylrhodiumdichloridu.
RhV
Sloučeniny RhV jsou obvykle nestabilní, ke stabilizaci se používají donorové ligandy – hydrid, silyl a boryl. Toto oxidační číslo se vyskytuje při borylacích.

Metalocykly
Cyklometalované sloučeniny rhodia mají významnou roli v organokovové chemii. Přestože jsou takové sloučeniny dobře popsané, tak se rhodité cyklometaláty obsahující azoskupiny neobjevují běžně. Jako příklad lze uvést hexakoordinovaný orthometalovaný rhoditý thiolátokomplex trans-[Rh(C∧N∧S)Cl(PPh3)2], připravený z benzyl 2-(fenylazo)fenylthioetheru a RhCl3·3H2O za přítomnosti nadbytku PPh3, přičemž dochází ke štěpení vazeb C(sp2)−H a C(sp3)−S. Jedná se o první známý případ komplexu (fenylazo)thioátového ligandu. Mechanismus tvorby orthometalovaných derivátů azobenzenu spočívá v počáteční koordinaci azodusíku a následné elektrofilní substituci na fenylové jádro. PPh3 je pro štěpení vazeb C(sp3)−S nezbytný. U vazeb C−S probíhá redukční štěpení jednoelektronovým mechanismem. Na rozdíl od odpovídajících (fenylazo)fenolátových sloučenin jsou reakce orthometalovaných thiolátových komplexů plně vratné, se standardním elektrodovým potenciálem 0,82 V oproti Ag/AgCl, což je způsobeno hlavně atomem síry v thiolátové skupině.[4]
Použití
I přes vysokou cenu se rhodium často používá jako katalyzátor.
Výroba kyseliny octové a jejího anhydridu
Monsanto proces je metoda průmyslové výroby kyseliny octové karbonylací methanolu,[5] který byl ovšem z velké části nahrazen na iridiu založeným Cativa procesem.
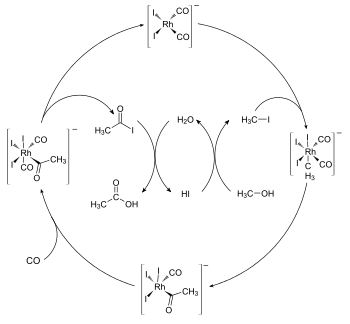
Aktivním katalyzátorem je zde anion cis-[Rh(CO)2I2]−,[6] který vstupuje do oxidační adice s jodmethanem. Podobný proces Tennessee-Eastman vytváří acetanhydrid karbonylací methylacetátu.[7]
- CH3CO2CH3 + CO → (CH3CO)2O
Hydroformylace
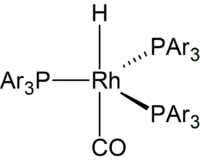
Při hydroformylacích se používají rhodiové katalyzátory; některé z nich jsou rozpustné ve vodě, což usnadňuje oddělení produktů od katalyzátoru.[8]

Hydrogenace
Wilkinsonův katalyzátor se používá jako homogenní katalyzátor hydrogenací alkenů.[9] Součástmi katalytického cyklu jsou oxidační adice vodíku, π-komplexace alkenu, migrační inserce (vnitromolekulární přesun hydridu nebo inserce alkenu) a redukční eliminace.
Kationtové organorhodné katalyzátory jsou vhodné pro asymetrické hydrogenace, používané na výrobu bioaktivních látek, například léčiv a agrochemikálií.[10]

Reference
V tomto článku byl použit překlad textu z článku Asymmetric hydrogenation na anglické Wikipedii.
- Synthesis of Organometallic Compounds: A Practical Guide Sanshiro Komiya Ed. S. Komiya, M. Hurano 1997
- CRABTREE, Robert H. The Organometallic Chemistry of the Transition Metals. 4th. vyd. USA: Wiley-Interscience, 2005. ISBN 0-471-66256-9. (anglicky)
- Bradford B. Wayland, Sujuan Ba, Alan E. Sherry. Activation of Methane and Toluene by Rhodium(II) Porphyrin Complexes. J. Am. Chem. Soc.. 1991, s. 5305–5311. DOI 10.1021/ja00014a025. (anglicky)
- K. Pramanik; U. DAS; B. ADHIKARI; D. CHOPRA; H. STOECKLI-EVANS. RhCl3-Assisted C-H and C-S Bond Scissions: Isomeric Self-Association of Organorhodium(III) Thiolato Complex. Synthesis, Structure, and Electrochemistry. Inorganic Chemistry. 2008, s. 429–438. DOI 10.1021/ic7016006. PMID 18161963. (anglicky)
- Hosea Cheung, Robin S. Tanke, G. Paul Torrence "Acetic Acid" in Ullmann's Encyclopedia of Industrial Chemistry, 2002, Wiley-VCH, Weinheim. DOI:10.1002/14356007.a01_045
- Hartwig, J. F. Organotransition Metal Chemistry, from Bonding to Catalysis; University Science Books: New York, 2010. ISBN 189138953X
- Zoeller, J. R.; AGREDA, V. H.; COOK, S. L.; LAFFERTY, N. L.; POLICHNOWSKI, S. W.; POND, D. M. Eastman Chemical Company Acetic Anhydride Process. Catalysis Today. 1992, s. 73–91. DOI 10.1016/0920-5861(92)80188-S. (anglicky)
- Cornils, B.; Herrmann, W. A. (eds.) “Aqueous-Phase Organometallic Catalysis” VCH, Weinheim: 1998
- HARTWIG, John F. Organotransition metal chemistry- From bonding to Catalysis. [s.l.]: University Science Books, 2010. ISBN 978-1-891389-53-5. (anglicky)
- KNOWLES, William S. Asymmetric Hydrogenations (Nobel Lecture). Angewandte Chemie International Edition. 2002, s. 1998. DOI 10.1002/1521-3773(20020617)41:12<1998::AID-ANIE1998>3.0.CO;2-8. (anglicky)
- H.-J.Drexler, Songlin Zhang, Ailing Sun, A. Spannenberg, A. Arrieta, A. Preetz, D. Heller. Cationic Rh-bisphosphine-diolefin complexes as precatalysts for enantioselective catalysis––what information do single crystal structures contain regarding product chirality?. Tetrahedron: Asymmetry. S. 2139–50. DOI 10.1016/j.tetasy.2004.06.036. (anglicky)