Asymetrická syntéza
Asymetrická syntéza , také nazývaná enantioselektivní syntéza, je druh chemické syntézy. IUPAC ji definuje jako chemickou reakci (nebo řadu reakcí), v níž je ve výchozí látce obsaženo jedno nebo více center chirality a při níž vznikají stereoizomerní (enantiomerní nebo diastereomerní) produkty v rozdílných množstvích. Zjednodušeně jde tedy o metodu syntézy, při níž je upřednostňován vznik určitého enantiomeru nebo diastereomeru.
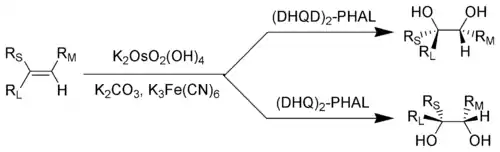
RL = největší substituent; RM = středně velký substituent; RS = nejmenší substituent
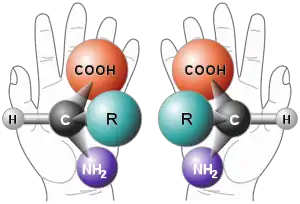
Enantioselektivní syntéza je důležitým procesem v moderní chemii a je zvlášť důležitá při výrobě léčiv, jelikož různé enantiomery či diastereomery často mají různou biologickou aktivitu.
Přehled
Mnoho stavebních prvků biologických systémů jako jsou sacharidy a aminokyseliny se v organismech tvoří pouze v jedom enantiomeru. Organismy tak mají vysokou míru chemické chirality a často různě reagují na odlišné izomery dané sloučeniny. Příklady této selektivity jsou:
- Chuť: umělé sladidlo aspartam má dva enantiomery. L-aspartam má sladkou chuť, zatímco D-aspartam je bez chuti.[1]
- Vůně: R (–) karvon má podobnou vůni jako máta klasnatá, S (+) karvon voní jako kmín kořenný.[2]
- Účinnost léčiv: antidepresivum Citalopram se prodává jako racemická směs. Bylo ovšem zjištěno, že pouze S (+) enantiomer má požadované účinky.[3][4]
- Bezpečnost léčiv: D‑penicilamin se využívá v chelační terapii a k léčbě revmatoidní artritidy. L‑penicilamin je toxický, jelikož brání správné funkci pyridoxinu.[5]
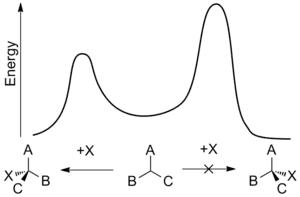
Některé enantioselektivní syntézy mají velký význam avšak je obtížné takové syntézy dosáhnout. Enantiomery mají shodné hodnoty standardní slučovací entalpie a entropie a tak by se v neřízených procesech měly tvořit ve stejném množství – za vzniku racemické směsi. Enantiomerní syntézu lze provést za použití chirální látky, která v přechodném stavu upřednostňuje tvorbu jednoho izomeru před tvorbou jiného. Toto ovlivňování se nazývá asymetrická indukce a příslušná chirální látka může být substrát, reaktant nebo katalyzátor a může také pocházet z okolního prostředí; funguje tak, že sníží aktivační energii potřebnou pro tvorbu jednoho z enantiomerů.
Enantioselektivita se často popisuje poměry rychlostí enantiodiferenciačních kroků—okamžiků, kdy se reaktant přemění v jeden z enantiomerních produktů. Rychlostní konstanta k dané reakce je funkcí její aktivační energie, občas nazývané energetická nebo aktivační bariéra, a je závislá na teplotě. Za použití Gibbsovy volné energie energetické bariéry, ΔG*, je relativní rychlost vzniku jednotlivých enantiomerů při teplotě T následující:

Tato teplotní závislost vytváří rozdíl v rychlostech příslušných reakcí a enantioselektivita je tedy při nižší teplotě větší. I malé rozdíly v aktivační energii mohou mít vést k zaznamenatelnému výsledku.
| ΔΔG* (kcal) | k1/k2 (273 K) | k1/k2 (298 K) | k1/k2 (323 K) |
|---|---|---|---|
| 1,0 | 6,37 | 5,46 | 4,78 |
| 2,0 | 40,6 | 29,8 | 22,9 |
| 3,0 | 259 | 162 | 109 |
| 4,0 | 1650 | 886 | 524 |
| 5,0 | 10 500 | 4830 | 2510 |
Možnosti asymetrické syntézy
Enantioselektivní katalyzátory
Enantioselektivní katalyzátory (tradičně označované jako asymetrické katalyzátory) jsou obecně chirální komplexní sloučeniny. Katalýza je účinná u širší skupiny reakcí než jakákoliv jiná metoda enantioselektivní syntézy. Většina enantioselektivních katalyzátorů má největší účinnost při malých hodnotách poměru substrát/katalyzátor.[6][7] Díky jejich vysoké účinnosti jsou často vhodné pro syntézy v průmyslovém měřítku i když jsou drahé.[8] Příkladem syntézy s použitím enantioselektivních katalyzátorů je asymetrická hydrogenace, která se používá k redukci řady funkčních skupin.
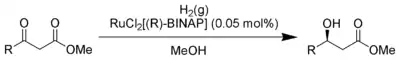
Ve vývoji nových katalyzátorů výrazně převažuje vývoj nových tříd ligandů. Některé ligandy, často nazývané „privilegované ligandy“, jsou účinné u velkého počtu reakcí; příklady takových ligandů jsou BINOL, Salen a bisoxazolinové ligandy. Celkově ovšem existuje jen několik málo katalyzátorů, které lze použít u více než jednoho typu asymetrických reakcí; například Nojoriho asymetrická hydrogenace s BINAP/Ru vyžaduje β-keton, zatímco jiný katalyzátor, BINAP/diamin-Ru, funguje rovněž s α,β-alkeny a aromatickými sloučeninami.
Chirální pomocníci
Chirální pomocník je organická sloučenina, která se spáruje s výchozí látkou za vzniku nové sloučeniny, která může projít enantioselektivními reakcemi přes vnitromolekulární asymetrickou indukci..[9][10] Na konci reakce je pomocník odstraněn za podmínek, které nevyvolávají racemizaci produktu[11] a obvykle je pro další použití obnoven.
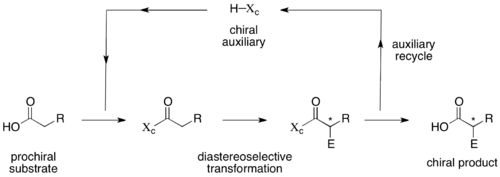
Chirální pomocníky je třeba, aby byly účinné, používat ve stechiometrickém množství a vyžadují navíc další kroky pro připojení a následné odstranění. V některých příkladech jsou však na nich založené jediné dostupné stereoselektivní metody a tyto reakce jsou univerzální a dobře prozkoumané, což umožňuje použít nejúčinnější způsob získání enantiomerně čistých produktů.[10] Produkty těchto reakcí jsou navíc diastereomery, díky čemuž je lze snadno oddělit metodami jako jsou sloupcová chromatografie nebo krystalizace.
Biokatalýza
Při biokatalýze se používají biomolekuly (konkrétně izolované enzymy) nebo živé buňky k provádění chemických reakcí. Výhody těchto reaktantů spočívají ve velmi vysoké enantiomerové a reaktantové selektivitě, stejně jako v mírných reakčních podmínkách a minimálním dopadu na životní prostředí. Biokatalyzátory se častěji používají v průmyslu než v akademickém výzkumu;[12] například při výrobě statinů.[13] Vysoká specifita reaktantu může být problematická a často je třeba vyzkoušet mnoho biokatalyzátorů, než je nalezen nejúčinnější.
Enantioselektivní organokatalýza
Organokatalýza je druh katalýzy, kdy se rychlost reakce zvyšuje působením organických sloučenin obsahujících uhlík, vodík, síru a/nebo jiné nekovové prvky. [14][15] Je-li organický katalyzátor chirální, může být dosaženo enantioselektivní syntézy,[16][17] například mnoho reakcí vytvářejících vazbu uhlík-uhlík se stává enantioselektivními za přítomnosti prolinu; takovou reakcí je mimo jiné aldolová reakce.[18]
V organokatalýze se jako chirální katalyzátory často využívají přírodní sloučeniny a sekundární aminy,[19] které nejsou drahé a jsou šetrné k životnímu prostředí, protože neobsahují kovy.
Alternativní metody
Alternativami enantioselektivní syntézy často zahrnují izolaci jednoho enantiomeru z racemické směsi některou z mnoha metod. V případech, kdy jsou levnější a méně časově náročné (nebo existuje využití pro oba enantiomery), může být tento postup výhodnější.
Oddělení a analýza enantiomerů
Dva enantiomery téže látky mají kromě optické otáčivosti stejné fyzikální vlastnosti jako jsou teploty tání a varu a polarita. To způsobuje, že se v chromatografii na tenké vrstvě pohybují se stejným Rf a mají stejné retenční časy v vysokoúčinné kapalinové chromatografii (HPLC) a plynové chromatografii (GC). Shodují se také jejich NMR a infračervená spektra.
Z těchto důvodů může být velmi obtížné určit, jaký proces vytváří jediný enantiomer (a o který z možných jde) stejně jako oddělit tyto enantiomery z produktů reakce, která není zcela enantioselektivní. Enantiomery téže sloučeniny však mají rozdílné chování v přítomnosti další chirální látky a toho lze využít k jejich oddělení a analýze.
Enantiomery mají rozdílný pohyb na chirálních médiích jako jsou křemen nebo standardní média, která byla chirálně modifikována. Tato skutečnost je základem chirální sloupcové chromatografie, kterou lze v malém měřítku použít k oddělení pomocí plynové chromatografie nebo HPLC a ve velkém měřítku na rozdělení chirálně nečistých materiálů. Tato metoda však vyžaduje velká množství dalších chirálních látek, což může být nákladné. Běžnou alternativou je použití chirálních derivačních činidel k přeměne enantiomerů na diastereomery, v podstatě stejným způsobem jako při použití chirálních pomocníků. Diastereomery mají rozdílné fyzikální vlastnosti a mohou tak být odděleny a analyzovány běžnými metodami. Zvláštním druhem chirálních derivačních činidel jsou chirální oddělovací činidla, která se používají v NMR spektroskopii stereoizomerů, často fungují na principu tvorby chirálních komplexů se sloučeninami europia jako jsou Eu(fod)3 a Eu(hfc)3.
Převaha jednoho enantiomeru látky může být rovněž určena za použití některých optických metod. Nejstarší takovou metodou je použití polarimetru ke srovnání optické otáčivosti dané směsi s jinou směsí o známém složení. Rovněž je možné použít UV-VIS spektroskopii stereoizomerů s využitím Cottonova jevu.
Jednou z nejpřesnějších metod určení chirality sloučeniny je určení její absolutní konfigurace pomocí rentgenové krystalografie, přičemž je však potřeba připravit vhodný monokrystal.
Historie
Začátky (1815–1905)
Roku 1815 ukázal francouzský lékař Jean-Baptiste Biot, že některé látky mohou stáčet rovinu polarizovaného světla (tato vlastnost se nazývá optická aktivita).[20] Příčina tohoto jevu nebyla známa do roku 1848, kdy Louis Pasteur navrhl, že má molekulární základ vycházející z určité formy asymetrie; označení chiralita zavedl William Thomson o rok později.[21]
Samotný původ chirality byl plně popsán v roce 1874, kdy Jacobus Henricus van 't Hoff a Joseph Le Bel nezávisle na sobě navrhli čtyřstěnnou geometrii uhlíku.[22][23] Předchozí strukturní modely byly dvourozměrné a van 't Hoff s Le Belem předpokládali, že uspořádání skupin kolem tohoto čtyřstěnu by mohlo určovat optickou aktivitu sloučeniny, což se dnes nazývá Le Belovo–van't Hoffovo pravidlo.
Roku 1894 Hermann Emil Fischer vytvořil pojem asymetrická indukce,[25] jímž popsal jako selektivní tvorbu D-glukózy v rostlinách jako ovlivněnou opticky aktivními složkami v chlorofylu. Rovněž sám úspěšně provedl první enantioselektivní syntézu, pomocí enantioselektivně prodlužovaných sacharidů (nazvanou Kilianiho–Fischerova syntéza).[26]
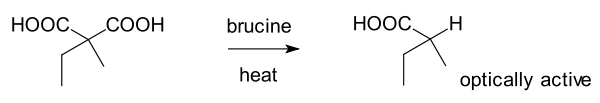
První enantioselektivní syntéza je nejčastěji připisována Willymu Marckwaldovi z Humboldtovy univerzity v Berlíně, jde o brucinem katalyzovanou enantioselektivní dekarboxylaci kyseliny 2-ethyl-2-methylmalonové popsanou roku 1904.[24][27] Při reakci vzniká mírný o něco více (−) formy vzniklého produktu, kyseliny 2-methylmáselné, než (+) formy; tato látka je rovněž přirozenou součástí postranního řetězce lovastatinu vytvářenou příslušnou diketidsyntázou (LovF) v průběhu jeho biosyntézy[28] - jedná se tedy o první zanamenanou enantioselektivní úplnou syntézu („první příklad asymetrické katalýzy, enantiotopního výběru a organokatalýzy“)[24] Tato reakce má rovněž historický význam, jelikož v té době nemohla být enantioselektivní syntéza vysvětlena pouze pomocí vitalismu. Mnoho významných chemiků daného období, jako například Jöns Jacob Berzelius, se domnívalo, že přírodní a umělé sloučeniny jsou zásadně odlišné a že chiralita je jednoduše projevem „životní síly“, která se vyskytuje pouze v přírodních sloučeninách. Marckwald na rozdíl od Fischera provedl enantioselektivní reakci s nechirální, „nepřirozenou“ výchozí látkou, i když s chirálním organickým katalyzátorem.[24]
Začátek rozvoje (1905–1965)
Rozvoj enantioselektivní syntézy byl ze začátku pomalý, hlavně kvůli nedostatku dostupných separačních a analytických metod. Diastereomery mají rozdílné fyzikální vlastnosti, což umožňuje oddělení konvenčními postupy, ovšem enantiometry bylo v té době možné oddělit pouze pomocí spontánního rozlišení (kde se enantiomery oddělují během krystalizace) nebo kinetického rozlišení (v tomto případě je jeden z enantiomerů selektivně zničen). Jedinou metodu analýzy enantiomerů tehdy představovalo měření optické aktivity polarimetrem, touto metodou nelze získat údaje o struktuře látky.
Pokrok v této oblasti začal v 50. letech 20. století díky chemikům jako byli Robert Burns Woodward a Vladimir Prelog, ale také díky rozvoji nových technik. První z nich byla rentgenová krystalografie, která byla v roce 1951 poprvé (Johannesem Bijvoetem) použita k určení absolutní konfigurace organické sloučeniny.[29]
O rok později použil Dalgliesh chirální chromatografii, zde konkrétně papírovou chromatografii, k oddělení chirálních aminokyselin.[30] I když nebyl prvním, kdo podobnou separaci provedl, tak správně přiřadil separaci enantiomerů k diferenciálnímu zadržování chirální celulózy. Tato metoda byla vylepšena roku 1960, kdy Klem a Reed popsali použití chirálně pozměněného silikagelu k chirální HPLC separaci.[31]
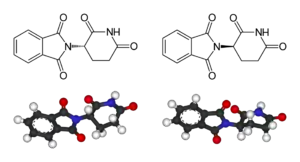
Levotočivý: (S)-thalidomid
Pravotočivý: (R)-thalidomid
Thalidomid
I když již bylo známo, že různé enantiomery téhož léčiva mohou mít rozdílné účinky, díky výzkumu, který provedl Arthur Robertson Cushny,[32][33] tak se s tímto při prvních návrzích a testech léčiv nepočítalo, po událostech kolem thalidomidu se vývoj a schvalování léčiv značně změnily.
Thalidomid, poprvé syntetizovaný v roce 1953, byl v letech 1957 až 1962 často předepisován k léčbě ranní nevolnosti, ale brzy bylo zjištěno, že je značně teratogenní,[34] když došlo k více než 10 000 vrozeným vývojovým vadám u novorozenců. V důsledku toho se v mnoha zemích zpřísnila pravidla pro testování a schvalování léčiv.
První výzkumy teratogenního mechanismu, prováděné na myších, ukázaly, že jeden izomer thalidomidu má teratogenní účinky, zatímco druhý má veškerou terapeutickou aktivitu. Tato domněnka byla později vyvrácena, ovšem vedla ke zvýšení významu chirality při vývoji léčiv, což vedlo k nárůstu výzkumu metod enantioselektivní syntézy.
Od roku 1965
Cahnova–Ingoldova–Prelogova pravidla priority, publikovaná v roce 1966, umožnila snazší a přesnější popis enantiomerů.[35][36] Ve stejném roce byla provedeno první úspěšné oddělení enantiomerů plynovou chromatografií,[37] což bylo důležité při rozvoji tehdy používané technologie.
William Standish Knowles, Rjódži Nojori a Karl Barry Sharpless objevili kovy katalyzovanou enantioselektivní syntézu, za tento objev získali roku 2001 Nobelovu cenu za chemii. Knowles a Nojori v roce 1968 nezávisle na sobě objevili asymetrickou hydrogenaci. Knowles nahradil nechirální trifenylfosfinové ligandy ve Wilkinsonově katalyzátoru chirálními fosfanovými ligandy. Tyto experimentální katalyzátory byly zapojeny do reakce, v níž se objevil 15procentní přebytek jednoho z enantiomerů. Knowles byl rovněž prvním, kdo použil kovy katalyzovanou enantioselektivní syntézu v průmyslové výrobě; při práci pro Monsanto vyvinul enantioselektivní hydrogenační krok k výrobě L-DOPA za použití DIPAMPu jako katalyzátoru:[38][39][40]
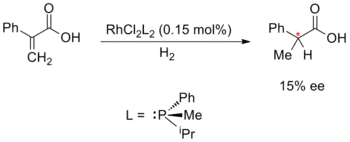 |
 | |
| Knowles: Asymetrická hydrogenace (1968) | Nojori: Enantioselekivní cyklopropanace (1968) |
|---|
Nojori vymyslel komplex mědi s chirální Schiffovou bází jako ligandem, který použil k mezimolekulární kovem katalyzované karbenoidové cyklopropanaci styrenu.[41] Společně s Knowlesovými poznatky byly Nojoriho hodnoty enantiomerního přebytku u těchto ligandů první generace nízké: pouze 6 %. Pokračující výzkum však vedl k rozvoji Nojoriho asymetrických hydrogenačních reakcí.
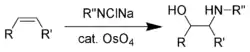
Sharpless během 70. a 80. let doplnil tyto redukční reakce vyvinutím řady asymetrických oxidací (Sharplessovy epoxidace,[42] asymetrické dihydroxylace[43] a oxyaminace).[44] První z těchto reakcí byla oxyaminace s použitím oxidu osmičelého.
Ve stejné době byly objeveny analýzy chirálních sloučenin pomocí NMR spektroskopie; v některých se používají chirální derivační činidla jako jsou Mosherova kyselina[45] nebo na europiu založené reaktanty, z nichž je první použitou látkou Eu(DPM)3.[46]
Používání chirálních pomocníků zavedl roku 1978 Elias James Corey.[47] Přibližně ve stejné době byla vyvinuta enantioselektivní organokatalýza, první práce na toto téma zmiňovaly mimo jiné Hajosovu–Parrishovu–Ederovu–Sauerovu–Wiechertovu reakci. Během 80. let se rozšířilo používání enzymaticky katalyzovaných reakcí,[48] a to především v průmyslu;[49] příkladem těchto reakcí je asymetrická hydrolýza esterů prasečí jaterní esterázou. Vznikající technologie genetického inženýrství umožňují přizpůsobení enzymů specifickým procesům a tím i více různých selektivních přeměn, například asymetrickou hydrogenaci prekurzorů statinů.[13]
Reference
V tomto článku byl použit překlad textu z článku Enantioselective synthesis na anglické Wikipedii.
- GAL, Joseph. The Discovery of Stereoselectivity at Biological Receptors: Arnaldo Piutti and the Taste of the Asparagine Enantiomers-History and Analysis on the 125th Anniversary. Chirality. 2012, s. 959–976. DOI 10.1002/chir.22071. PMID 23034823. (anglicky)
- Theodore J. Leitereg; DANTE G. GUADAGNI; JEAN HARRIS; THOMAS R. MON; ROY TERANISHI. Chemical and sensory data supporting the difference between the odors of the enantiomeric carvones. J. Agric. Food Chem.. 1971, s. 785–787. DOI 10.1021/jf60176a035. (anglicky)
- Lepola U, Wade A, Andersen HF. Do equivalent doses of escitalopram and citalopram have similar efficacy? A pooled analysis of two positive placebo-controlled studies in major depressive disorder. Int Clin Psychopharmacol. May 2004, s. 149–55. DOI 10.1097/01.yic.0000122862.35081.cd. PMID 15107657. (anglicky)
- HYTTEL, J.; BØGESØ, K. P.; PERREGAARD, J.; SÁNCHEZ, C. The pharmacological effect of citalopram resides in the (S)-(+)-enantiomer. Journal of Neural Transmission. 1992, s. 157–160. DOI 10.1007/BF01244820. PMID 1632943. (anglicky)
- JAFFE, IA; ALTMAN, K; MERRYMAN, P. The Antipyridoxine Effect of Penicillamine in Man.. The Journal of Clinical Investigation. Oct 1964, s. 1869–73. DOI 10.1172/JCI105060. PMID 14236210. (anglicky)
- N. JACOBSEN, Eric; PFALTZ, Andreas; YAMAMOTO, Hisashi. Comprehensive asymmetric catalysis 1-3. Berlin: Springer, 1999. ISBN 9783540643371. (anglicky)
- M. Heitbaum; F. GLORIUS; I. ESCHER. Asymmetric Heterogeneous Catalysis. Angewandte Chemie International Edition. 2006, s. 4732–4762. DOI 10.1002/anie.200504212. PMID 16802397. (anglicky)
- Asymmetric Catalysis on Industrial Scale, (Blaser, Schmidt), Wiley-VCH, 2004.
- ROOS, Gregory. Compendium of chiral auxiliary applications.. San Diego, Calif.: Acad. Press, 2002. ISBN 9780125953443. (anglicky)
- Glorius, F.; GNAS, Y. Chiral Auxiliaries – Principles and Recent Applications. Synthesis. 2006, s. 1899–1930. DOI 10.1055/s-2006-942399. (anglicky)
- EVANS, D. A.; HELMCHEN, G.; RÜPING, M. Asymmetric Synthesis – The Essentials. Redakce Christmann M.. [s.l.]: Wiley-VCH Verlag GmbH & Co., 2007. ISBN 978-3-527-31399-0. Kapitola Chiral Auxiliaries in Asymmetric Synthesis, s. 3–9. (anglicky)
- SCHMID, A.; DORDICK, J. S.; HAUER, B.; KIENER, A.; WUBBOLTS, M.; WITHOLT, B. Industrial biocatalysis today and tomorrow. Nature. 2001, s. 258–268. DOI 10.1038/35051736. PMID 11196655. (anglicky)
- MÜLLER, Michael. Chemoenzymatic Synthesis of Building Blocks for Statin Side Chains. Angewandte Chemie International Edition. 7 January 2005, s. 362–365. DOI 10.1002/anie.200460852. PMID 15593081. (anglicky)
- Berkessel, A.; GROEGER, H. Asymmetric Organocatalysis. Weinheim: Wiley-VCH, 2005. ISBN 3-527-30517-3. (anglicky)
- LIST, Benjamin. Organocatalysis. Chem. Rev.. 2007, s. 5413–5883. DOI 10.1021/cr078412e. (anglicky)
- GRÖGER, Albrecht Berkessel; Harald. Asymmetric organocatalysis – from biomimetic concepts to applications in asymmetric synthesis. 1. ed., 2. reprint.. vyd. Weinheim: Wiley-VCH, 2005. ISBN 3-527-30517-3. (anglicky)
- DALKO, Peter I.; MOISAN, LIONEL. Enantioselective Organocatalysis. Angewandte Chemie International Edition. 15 October 2001, s. 3726–3748. DOI 10.1002/1521-3773(20011015)40:20<3726::AID-ANIE3726>3.0.CO;2-D. (anglicky)
- NOTZ, Wolfgang; TANAKA, FUJIE; BARBAS, CARLOS F. Enamine-Based Organocatalysis with Proline and Diamines: The Development of Direct Catalytic Asymmetric Aldol, Mannich, Michael, and Diels−Alder Reactions. Accounts of Chemical Research. 1 August 2004, s. 580–591. DOI 10.1021/ar0300468. PMID 15311957. (anglicky)
- BERTELSEN, Søren; JØRGENSEN, Karl Anker. Organocatalysis—after the gold rush. Chemical Society Reviews. 2009, s. 2178–89. DOI 10.1039/b903816g. PMID 19623342. (anglicky)
- Lakhtakia, A. (ed.). Selected Papers on Natural Optical Activity (SPIE Milestone Volume 15). [s.l.]: SPIE, 1990. (anglicky)
- Pedro Cintas. Tracing the Origins and Evolution of Chirality and Handedness in Chemical Language. Angewandte Chemie International Edition. 2007, s. 4016–4024. DOI 10.1002/anie.200603714. PMID 17328087. (anglicky)
- LE BEL, Joseph. Sur les relations qui existent entre les formules atomiques des corps organiques et le pouvoir rotatoire de leurs dissolutions. Bull. Soc. Chim. Fr. 1874, s. 337-347. Dostupné online. (anglicky)
- van 't Hoff, J.H. (1874) "Sur les formules de structure dans l'espace" (On structural formulas in space), Archives Néerlandaises des Sciences Exactes et Naturelles, 9 : 445–454.
- KOSKINEN, Ari M.P. Asymmetric synthesis of natural products. Second. vyd. Hoboken, N.J.: Wiley, 2013. ISBN 1118347331. S. 17, 28–29. (anglicky)
- FISCHER, Emil. Synthesen in der Zuckergruppe II. Berichte der deutschen chemischen Gesellschaft. 1 October 1894, s. 3189–3232. DOI 10.1002/cber.189402703109. (anglicky)
- FISCHER, Emil; HIRSCHBERGER, JOSEF. Ueber Mannose. II. Berichte der deutschen chemischen Gesellschaft. 1 January 1889, s. 365–376. DOI 10.1002/cber.18890220183. (anglicky)
- Marckwald, W. Ueber asymmetrische Synthese. Berichte der deutschen chemischen Gesellschaft. 1904, s. 349–354. DOI 10.1002/cber.19040370165. (anglicky)
- CAMPBELL, Chantel D.; VEDERAS, John C. Biosynthesis of lovastatin and related metabolites formed by fungal iterative PKS enzymes. Biopolymers. 23 June 2010, s. 755–763. DOI 10.1002/bip.21428. (anglicky)
- BIJVOET, J. M.; PEERDEMAN, A. F.; VAN BOMMEL, A. J. Determination of the Absolute Configuration of Optically Active Compounds by Means of X-Rays. Nature. 1951, s. 271–272. DOI 10.1038/168271a0. Bibcode 1951Natur.168..271B. (anglicky)
- DALGLIESH, C. E. 756. The optical resolution of aromatic amino-acids on paper chromatograms. Journal of the Chemical Society (Resumed). 1952, s. 3940. DOI 10.1039/JR9520003940. (anglicky)
- KLEMM, L.H.; REED, DAVID. Optical resolution by molecular complexation chromatography. Journal of Chromatography A. 1960, s. 364–368. DOI 10.1016/S0021-9673(01)97011-6. (anglicky)
- CUSHNY, AR. Atropine and the hyoscyamines-a study of the action of optical isomers. The Journal of Physiology. 2 November 1903, s. 176–94. DOI 10.1113/jphysiol.1903.sp000988. PMID 16992694. (anglicky)
- CUSHNY, AR; PEEBLES, AR. The action of optical isomers: II. Hyoscines. The Journal of Physiology. 13 July 1905, s. 501–10. DOI 10.1113/jphysiol.1905.sp001097. PMID 16992790. (anglicky)
- MCBRIDE, W. G. Thalidomide and Congenital Abnormalities. The Lancet. 1961, s. 1358. DOI 10.1016/S0140-6736(61)90927-8. (anglicky)
- Robert Sidney Cahn; Christopher Kelk Ingold; Vladimir Prelog. Specification of Molecular Chirality. Angewandte Chemie International Edition. 1966, s. 385–415. DOI 10.1002/anie.196603851. (anglicky)
- Vladimir Prelog; Günter Helmchen. Basic Principles of the CIP-System and Proposals for a Revision. Angewandte Chemie International Edition. 1982, s. 567–583. DOI 10.1002/anie.198205671. (anglicky)
- GIL-AV, Emanuel; FEIBUSH, BINYAMIN; CHARLES-SIGLER, ROSITA. Separation of enantiomers by gas liquid chromatography with an optically active stationary phase. Tetrahedron Letters. 1966, s. 1009–1015. DOI 10.1016/S0040-4039(00)70231-0. (anglicky)
- VINEYARD, B. D.; KNOWLES, W. S.; SABACKY, M. J.; BACHMAN, G. L.; WEINKAUFF, D. J. Asymmetric hydrogenation. Rhodium chiral bisphosphine catalyst. Journal of the American Chemical Society. 1977, s. 5946–5952. DOI 10.1021/ja00460a018. (anglicky)
- KNOWLES, William S. Asymmetric Hydrogenations (Nobel Lecture). Angewandte Chemie International Edition. 2002, s. 1998. DOI 10.1002/1521-3773(20020617)41:12<1998::AID-ANIE1998>3.0.CO;2-8. (anglicky)
- KNOWLES, W. S. Application of organometallic catalysis to the commercial production of L-DOPA. Journal of Chemical Education. March 1986, s. 222. DOI 10.1021/ed063p222. Bibcode 1986JChEd..63..222K. (anglicky)
- H. Nozaki; H. TAKAYA; S. MORIUTI; R. NOYORI. Homogeneous catalysis in the decomposition of diazo compounds by copper chelates: Asymmetric carbenoid reactions. Tetrahedron. 1968, s. 3655–3669. DOI 10.1016/S0040-4020(01)91998-2. (anglicky)
- KATSUKI, Tsutomu; SHARPLESS, K. BARRY. The first practical method for asymmetric epoxidation. Journal of the American Chemical Society. 1980, s. 5974–5976. DOI 10.1021/ja00538a077. (anglicky)
- JACOBSEN, Eric N.; MARKO, ISTVAN.; MUNGALL, WILLIAM S.; SCHROEDER, GEORG.; SHARPLESS, K. BARRY. Asymmetric dihydroxylation via ligand-accelerated catalysis. Journal of the American Chemical Society. 1988, s. 1968–1970. DOI 10.1021/ja00214a053. (anglicky)
- SHARPLESS, K. Barry; PATRICK, DONALD W.; TRUESDALE, LARRY K.; BILLER, SCOTT A. New reaction. Stereospecific vicinal oxyamination of olefins by alkyl imido osmium compounds. Journal of the American Chemical Society. 1975, s. 2305–2307. DOI 10.1021/ja00841a071. (anglicky)
- J. A. Dale, D. L. Dull and H. S. Mosher. α-Methoxy-α-trifluoromethylphenylacetic acid, a versatile reagent for the determination of enantiomeric composition of alcohols and amines. J. Org. Chem.. 1969, s. 2543–2549. DOI 10.1021/jo01261a013. (anglicky)
- HINCKLEY, Conrad C. Paramagnetic shifts in solutions of cholesterol and the dipyridine adduct of trisdipivalomethanatoeuropium(III). A shift reagent. Journal of the American Chemical Society. 1969, s. 5160–5162. DOI 10.1021/ja01046a038. PMID 5798101. (anglicky)
- ENSLEY, Harry E.; PARNELL, CAROL A.; COREY, ELIAS J. Convenient synthesis of a highly efficient and recyclable chiral director for asymmetric induction. The Journal of Organic Chemistry. 1978, s. 1610–1612. DOI 10.1021/jo00402a037. (anglicky)
- SARIASLANI, F.Sima; ROSAZZA, JOHN P.N. Biocatalysis in natural products chemistry. Enzyme and Microbial Technology. 1984, s. 242–253. DOI 10.1016/0141-0229(84)90125-X. (anglicky)
- WANDREY, Christian; LIESE, ANDREAS; KIHUMBU, DAVID. Industrial Biocatalysis: Past, Present, and Future. Organic Process Research & Development. 2000, s. 286–290. DOI 10.1021/op990101l. (anglicky)