SN1 reakce
SN1 reakce je druh organické reakce, kde SN označuje nukleofilní substituci a číslo 1 naznačuje, že jde o jednomolekulární reakci.[1][2] Reakce se tedy popisuje jako prvního řádu vzhledem k elektrofilu a nultého řádu vzhledem k nukleofilu; tento popis je vhodný v případech, kdy je koncentrace nukleofilu výrazně větší než koncentrace meziproduktů. Meziproduktem bývá karbokation.
SN1 reakce probíhají u sekundárních a terciárních alkylhalogenidů, v silně zásaditém nebo silně kyselém prostředí, se sekundárními a terciárními alkoholy. U primárních a někdy také sekundárních alkylhalogenidů místo toho probíhá SN2 reakce. Anorganická obdoba SN1 reakce bývá často označována pojmem disociační mechanismus, ten lze dobře vysvětlit pomocí cis efektu. Mechanismus navrhl Christopher Ingold v roce 1940.[3] Průběh SN1 reakcí nebývá, na rozdíl od SN2 reakcí, ovlivňován sílou nukleofilu.
Mechanismus
K vysvětlení mechanismu SN1 reakcí lze použít například hydrolýzu terc-butylbromidu za vzniku terc-butanolu:

Tato reakce probíhá ve třech krocích:
- Tvorba terc-butylového karbokationtu oddělením odcházející skupiny (zde bromidového aniontu) od atomu uhlíku; tento krok probíhá nejpomaleji a je vratný.
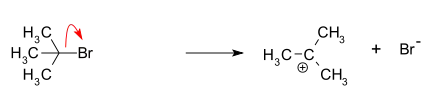
 Rekombinace karbokationtu s nukleofilem
Rekombinace karbokationtu s nukleofilem
- Nukleofilní atak: karbokation reaguje s nukleofilem. Pokud je nukleofilem neutrální molekula (například rozpouštědlo), je k dokončení reakce nutný třetí krok. Jestliže je rozpouštědlem voda, tak jako meziprodukt vzniká hydroniový ion. Tento krok probíhá rychle.

- Deprotonace: odštěpení protonu z protonovaného nukleofilu pomocí vody (která zde reaguje jako zásada) za tvorby alkoholu a hydroniového iontu. Tento krok probíhá rychle.
![]()
Vliv substituentů
SN1 mechanismus převažuje u reaktantů, které mají na centrální atom uhlíku navázané objemné funkční skupiny, jež stericky znesnadňují SN2 mechanismus. Takovéto substituenty rovněž zrychlují tvorbu karbokationtu, protože snižují vliv odpudivých van der Waalsových sil. Karbokation také stabilizují vlivem indukčních efektů i hyperkonjugace. SN1 mechanismus je hlavním mechanismem u reaktantů s terciárními alkylovými centry.
Příkladem takové reakce je příprava 2,5-dichlor-2,5-dimethylhexanu z odpovídajícího diolu pomocí koncentrované kyseliny chlorovodíkové:[4]
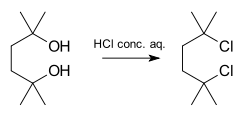
Při navyšování úrovně substituce v alfa a beta polohách vůči odcházejícím skupinám roste převaha SN1 mechanismu nad mechanismem SN2.
Stereochemie
Karbokation, který se vytváří v prvním kroku reakce, má sp2 hybridizovaný uhlík s trojúhelníkovou geometrií molekuly. Díky tomu může k nukleofilnímu ataku dojít ze dvou různých stran molekuly. Pokud není upřednostňován žádný z těchto směrů, tak, pokud reakce probíhá na chirálním centru, vzniká racemická směs.[5] Na níže uvedeném obrázku je tato skutečnost znázorněna na reakci S-3-chlor-3-methylhexanu s jodidovým iontem, vedoucí ke vzniku racemické směsi obou stereoizomerů 3-jod-3-methylhexanu:
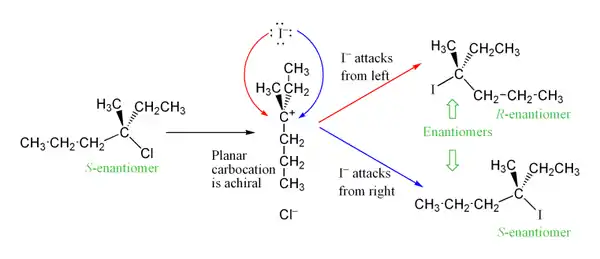
Pokud odcházející skupina zůstane v blízkosti meziproduktu krátkou dobu a uzavře cestu pro nukleofilní atak z jedné strany, tak se jeden ze stereoizomerů bude tvořit ve větším množství. Tímto se SN1 reakce liší od SN2 reakcí, které mají obrácený stereospecifický mechanismus, jelikož se nukleofil přibližuje k zadní straně odcházející skupiny.
Vedlejší reakce
Nejčastějšími vedlejšími reakcemi při jednomolekulárních nukleofilních substitucích jsou eliminační a přesmykové reakce. Pokud se substituce provádí za vyšších teplot, tak převažuje jednomolekulární (E1) eliminace, vedoucí k tvorbě alkenu. Při nižších teplotách probíhají obě reakce v podobné míře. K tvorbě alkenu dochází i při velmi nízkých reakčních teplotách. Pokusy o provedení SN1 reakce pomocí silně zásaditého nukleofilu, jako je například hydroxidový či methoxidový anion, pak bude vznikat alken E2 reakcí, obzvláště při vyšších teplotách. Pokud se karbokation může přesmyknout na stabilnější karbokation, tak se bude převážně tvořit produkt vzniklý z tohoto stabilnějšího karbokationtu.
Vliv rozpouštědla
Jelikož při SN1 reakci vzniká karbokationtový meziprodukt v nejpomalejším kroku (který tedy určuje rychlost reakce), tak lze samotnou reakci urychlit jakýmkoliv zásahem, který urychlí tvorbu karbokationtu. Běžně se používají jak aprotická (která stabilizují iontové meziprodukty) a protická (která částečně solvatují odcházející skupinu) rozpouštědla. Jako protická rozpouštědla často slouží voda nebo alkoholy, které také působí jako nukleofily a způsobují solvolýzu.
K porovnávání rychlosti solvolýzy vyvolávané různými rozpouštědly se používá Y-ová stupnice, jež porovnává dané rozpouštědlo (k) se směsí ethanolu (80 % objemových) a vody (20 % objemových) (k0) pomocí následujícího vzorce:

kde m je konstanta reaktantu (u terc-butylchloridu je rovna 1) a Y je parametr rozpouštědla.[6] Čistý ethanol má Y = −2,3, 50% ethanol má Y = 1,65 a u 15%ethanolu je Y = 3,2.[7]
Odkazy
Související články
Externí odkazy
 Obrázky, zvuky či videa k tématu SN1 reakce na Wikimedia Commons
Obrázky, zvuky či videa k tématu SN1 reakce na Wikimedia Commons
Reference
V tomto článku byl použit překlad textu z článku SN1 reaction na anglické Wikipedii.
- L. G. Wade, Jr., Organic Chemistry, 6th ed., Pearson/Prentice Hall, Upper Saddle River, New Jersey, USA, 2005
- J. March. Advanced Organic Chemistry. New York: Wiley, 1992. (4). ISBN 0-471-60180-2.
- L. C. Bateman; M. G. Church; E. D. Hughes; C. K. Ingold; N. A. Taher. 188. Mechanism of substitution at a saturated carbon atom. Part XXIII. A kinetic demonstration of the unimolecular solvolysis of alkyl halides. (Section E) a general discussion. Journal of the Chemical Society (Resumed). 1940, s. 979. DOI 10.1039/JR9400000979.
- Carl E. Wagner; Pamela A. Marshall. Synthesis of 2,5-Dichloro-2,5-dimethylhexane by an SN1 Reaction. Journal of Chemical Education. 2010, s. 81–83. DOI 10.1021/ed8000057. Bibcode 2010JChEd..87...81W.
- Sorrell, Thomas N. "Organic Chemistry, 2nd Edition" University Science Books, 2006
- Ernest Grunwald; S. Winstein. The Correlation of Solvolysis Rates. Journal of the American Chemical Society. 1948, s. 846. DOI 10.1021/ja01182a117.
- Arnold H. Fainberg; S. Winstein. Correlation of Solvolysis Rates. III.1 t-Butyl Chloride in a Wide Range of Solvent Mixtures. Journal of the American Chemical Society. 1956, s. 2770. DOI 10.1021/ja01593a033.