Fotoredoxní katalýza
Fotoredoxní katalýza je odvětví fotochemie, které se zabývá reakcemi, při nichž dochází k přenosu jednoho elektronu. Fotoredoxní katalyzátory se obvykle dělí na tři skupiny: komplexy přechodných kovů, organická barviva a polovodiče. V 90. letech 20. století se zde používaly především organické katalyzátory,[1] později se začaly více používat komplexy přechodných kovů.
_Schematic.png.webp)
Fotochemie senzitizátorů
Sensitizátory absorbují světlo a vytvářejí tím redoxně aktivní excitované stavy. U mnoha sensitizátorů založených na kovech se excitace dosahuje přenosem náboje z kovu na ligand, kdy se elektron přesune z kovu (například orbitalu d) na orbital ligandu (například π* orbital aromatického ligandu). Původní excitovaný elektronový stav se dostane do singletového excitovaného stavu vnitřní konverzí, stavu, kdy je energie rozptýlena spíše jako vibrační než jako energie elektromagnetického záření. Tento singletový excitovaný stav se může dále uvolňovat dvěma různými procesy: katalyzátor může fluoreskovat, kdy vyzáří foton a vrátí se do singletového základního stavu, nebo se přeměnit do energeticky nejnižšího tripletového excitovaného stavu (kdy dva nespárované elektrony mají stejný spin).
Přímý přechod z excitovaného tripletu do základního stavu, fosforescence, vyžaduje jak vyzáření fotonu, tak i obrácení spinu excitovaného elektronu. Tento mechanismus je pomalý, protože je spinově nedovolený, což prodlužuje životnost tripletového stavu. U běžného fotosenzitizátoru tris-(2,2’-bipyridyl)chloridu ruthenatého (zkráceně [Ru(bipy)3]2+ nebo [Ru(bpy)3]2+) jde o přibližně 1 100 ns. Tento čas je dostatečně dlouhý na to, aby mohlo k ostatním druhům uvolnění dojít před přeměnou katalyzátoru do základního stavu.
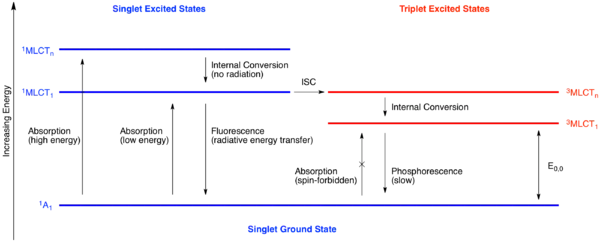
Dlouhotrvající tripletový excitovaný stav získatelný fotoexcitací je jak lepším redukčním činidlem, tak i lepším oxidačním činidlem než základní stav katalyzátoru. Jelikož je senzitizátor koordinačně nasycený, tak musí přesun elektronu proběhnout ve vnější sféře, kde se elektron protuneluje od katalyzátoru k substrátu.
Přesun elektronu ve vnější sféře
Marcusova teorie přesunu elektronu ve vnější sféře předpovídá, že kvantové tunelování bude nejrychlejší v systémech, ve kterých je přesun elektronu termodynamicky výhodný (například mezi silným redukčním a oxidačním činidlem) a kde je energetická bariéra přesunu nízká.
Energetická bariéra se odvozuje z Franckova–Condonova principu, kdy se předpokládá, že přesun elektronů proběhne rychleji při větším překryvu původního a výsledného elektronového stavu. Velikost této bariéry souvisí s mírou náchylnosti systému k přeuspořádání, tedy s rozdílem mezi vlnovými funkcemi původního a výsledného stavu elektronu.
Při vnitromolekulárním přesunu má podobný význam náchylnost jader k přesunu v závislosti na změně elektronového prostředí. Ihned po přesunu elektronu je jaderné uspořádání molekuly, do té doby rovnovážné, tvořené vibračně excitovaným stavem a musí se dostat do nového rovnovážného stavu. Systémy, jejichž energie příliš nezáleží na oxidačním čísle, tak vykazují menší vibrační excitace a nižší energetické bariéry. Fotokatalyzátory jako je [Ru(bipy)3]2+, mají pevné uspořádání, kde bidentátní ligandy zaujímají oktaedrickou geometrii okolo kovového centra. U komplexu tak v průběhu přesunu elektronu nedochází k výrazné reorganizaci.
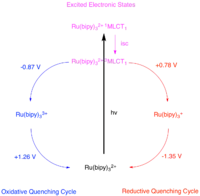
Obnovení katalyzátoru
Aby došlo k návratu do základního stavu, tak se katalyzátor musí zapojit do dalšího přesunu elektronu ve vnější sféře. V řadě případů tento proces probíhá za přítomnosti stechiometrického množství dvouelektronové redukčního nebo oxidačního činidlo, někdy se však do něj zapojuje další reaktant.
Protože část katalytického cyklu, při které nastává přesun elektronu, probíhá u tripletového excitovaného stavu, tak probíhá současně s fosforescenčním procesem. Pomocí Sternových–Volmerových experimentů je možné změřit intenzitu fosforescence změnami koncentrací jednotlivých možných reaktantů. Když se mění koncentrace pravého reaktantu, tak dochází ke změnám v rychlosti přesunu elektronů a intenzitě fosforescence. Tento vztah lze vyjádřit následující rovnicí:

kde I0 a I jsou intenzity vyzařování za přítomnosti a nepřítomnosti činidla, kq je rychlostní konstanta procesu, τ0 doba života excitovaného stavu za nepřítomnosti činidla a [Q] koncentrace reaktantu. Je-li tak známa doba života excitovaného stavu fotoredoxního katalyzátoru, tak lze určit rychlostní konstantu za přítomnosti jedné reagující látky měřením změn intenzity záření v závislosti na změnách koncentrace činidla.
Fotofyzikální vlastnosti
Redoxní potenciály
Redoxní potenciály fotoredoxních katalyzátorů musí být v souladu s potenciály reakcí ostatních látek. Zatímco redoxní potenciály základních stavů je možné snadno změřit cyklickou voltametrií nebo jinými elektrochemickými metodami, tak přímé měření redoxních potenciálů excitovaných stavů těmito postupy není možné;[2] jsou však známy dva postupy umožňující odhad redoxních potenciálů excitovaných stavů a jedna metoda pro jejich přímé měření. U odhadování potenciálů se při jednom postupu srovnávají rychlosti přesunu elektronů z excitovaného stavu pomocí souboru několika reaktantů v základních stavech, jejichž redoxní potenciály jsou známy. Častější je využití Rehmovy-Wellerovy rovnice, jež popisuje potenciály excitovaných stavů jako opravy potenciálů základních stavů:


, kde E*1/2 je redukční či oxidační potenciál excitovaného stavu, E1/2 redukční či oxidační potenciál základního stavu, E0,0 rozdíl energií mezi nultými vibračními stavy základního a excitovaného stavu a wr funkce vyjadřující elektrostatické interakce vznikající v důsledku oddělení nábojů během přesunu elektronu. E0,0 se přibližně vyjadřuje pomocí odpovídajícího přechodu ve fluorescenčním spektru. Tento postup umožňuje určit redoxní potenciály excitovaných stavů ze snadno změřitelných redoxních potenciálů základních stavů a spektroskopických dat.
Přímé měření redoxních potenciálů excitovaných stavů lze provést fázově modulovanou voltametrií. Tato metoda spočívá v ozáření elektrochemického článku světlem k získání části v excitovaných stavech, ovšem se sinusoidovitým řízením intenzity světla, díky čemuž koncentrace excitovaných stavů není stálá; měla by se měnit v souladu s intenzitou přicházejícího záření. Pokud je potenciál přivedený na článek dostatečně vysoký k vyvolání přenosu elektronů, tak lze změny koncentrace excitovaných stavů zodpovědných za redoxní reakci měřit jako střídavý proud. Fázový posun tohoto střídavého proudu v závislosti na intenzitě záření odpovídá průměrné životnosti excitovaných stavů před přesunem elektronů.
Existují tabulky redoxních potenciálů pro nejčastěji používané fotoredoxní katalyzátory.[3]
Elektronegativita ligandů
Relativní oxidační a redukční vlastnosti fotokatalyzátorů lze pochopit při využití elektronegativity ligandů a kovu tvořícího centrum komplexu. Elektronegativnější kovy a ligandy stabilizují elektrony lépe než ty, jejichž elektronegativa je nižší. Komplexy s elektronegativnějšími ligandy jsou tak více oxidující než ty s elektropozitivnějšími ligandy. Jako příklad lze uvést 2,2'-bipyridin a 2,2'-fenylpyridin, které jsou izoelektronické, co se týká jejich struktury, jelikož mají stejný počet a uspořádání elektronů. Fenylpyridin má oproti bypiridinu jeden atom dusíku nahrazený uhlíkem. Uhlík má menší elektronegativitu než dusík, takže elektrony zadržuje slaběji. Protože je zbylá část molekul ligandů stejná a fenylpyridin zadržuje elektrony slaběji než bipyridin, tak je méně elektronegativním ligandem a komplexy obsahující fenylpyridin jsou silněji redukující a slaběji oxidující než odpovídající komplexy s bipyridinovými ligandy.
Podobně je fluorovaný fenylpyridin elektronegativnější než fenylpyridin, takže komplexy obsahující fluorované ligandy jsou silnějšími oxidanty a slabšími reduktanty než příslušné nesubstituované fenylpyridinové komplexy. Vliv kovového centra na elektronové vlastnosti komplexu zahrnuje i jiné jevy. Paulingova elektronegativita ruthenia i iridia má hodnotu 2,2. Pokud by elektronegativita byla jediným faktorem ovlivňujícím redoxní potenciály, tak by komplexy ruthenia a iridia se stejnými ligandy měly být stejně silnými fotoredoxními katalyzátory. Podle Rehmovy-Wellerpvy rovnice mají však také spektroskopické vlastnosti vliv na redoxní vlastnosti excitovaných stavů.[4]
Parametr E0,0 souvisí s vlnovou délkou záření vydávaného komplexem a tak i, v závislosti na velikosti Stokesova posunu, rozdíl v energiích maximální absorpce a emise u dané molekuly. Komplexy ruthenia obecně mívají velké Stokesovy posuny a tak vyzařují záření o nižších energiích, tedy delších vlnových délkách, a mají malé excitační energie ve srovnání s komplexy iridia. Zatímco základní stavy komplexů obsahujících ruthenium mohou být dobrými redukčními činidly, tak jejich excitované stavy jsou slabšími redukčními i oxidačními činidly než odpovídající komplexy obsahující iridium. Díky tomu se iridium při vývoji katalyzátorů organických reakcí používá častěji, protože silnější redoxní potenciály excitovaných katalyzátorů umožňují použití slabších stechiometrických reduktantů a oxidantů, případně méně reaktivních substrátů.[4]
Použití
Reduktivní dehalogenace
První využití fotoredoxní katalýzy při reduktivních dehalogenacích byla omezena malým počtem využitelných substrátů a vedlejšími reakcemi.[5]
Neaktivované vazby uhlík-jod mohou být redukovány s využitím silného redukčního fotokatalyzátoru tris-(2,2’-fenylpyridin)iridia (Ir(ppy)3).[6] Vyšší redukční potenciál Ir(ppy)3 oproti [Ru(bipy)3]2+ umožňuje přímou redukci vazeb uhlík-jod bez použití stechiometrického redukčního činidla. Komplex iridia přenáší elektron na substrát, což vyvolává fragmentaci substrátu a oxidaci iridia na oxidační číslo IV. Oxidovaný fotokatalyzátor se navrací do původního oxidačního stavu oxidací reakčních příměsí.
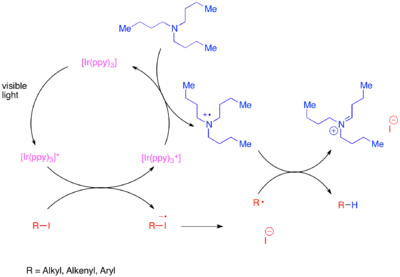
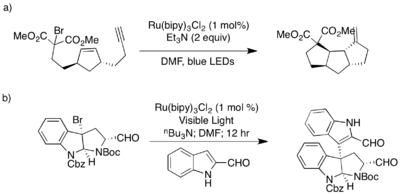
Podobně jako u cínem řízených radikálových dehalogenací mohou fotokatalytické reduktivní dehalogenace spouštět kaskádové cyklizace.[7]
Oxidativní tvorba iminiových iontů
Iminové ionty jsou dobrými elektrofily použitelnými k tvorbě vazeb C-C. Kondenzace aminů s karbonylovými sloučeninami za vzniku iminiových iontů je často nevýhodná a je třeba ji provádět za tvrdých dehydratujících podmínek. Byly tak vyvinuty jiné metody, založené na oxidaci příslušných aminů. Iminiové ionty je možné získat z aktivovaných aminů pomocí Ir(dtbbpy)(ppy)2PF6 jako fotoredoxního katalyzátoru.[8]
Při této přeměně pravděpodobně dochází k oxidaci aminu na aminiový radikálový kation excitovaným fotokatalyzátorem. Následně dojde k přesunu atomu vodíku na nadstechiometrický oxidant, například trichlormethylový radikál (CCl3), za vzniku iminiového iontu. Iminiový ion poté reaguje s nukleofilem. Byly také prozkoumány podobné reakce aminů s mnoha jinými nukleofily, například kyanidy (Streckerova syntéza aminokyselin), silylenolethery (Mannichova reakce), dialkylfosfáty, allylsilany (aza-Sakuraiova reakce), indoly (Friedelova–Craftsova reakce) a acetylidy mědi.[9][10][11][12][13]
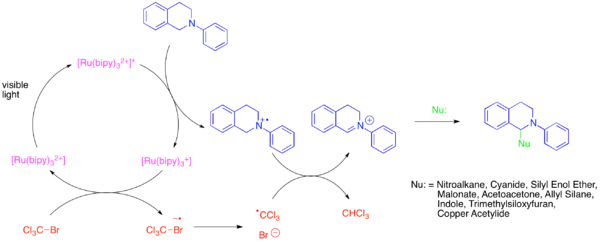
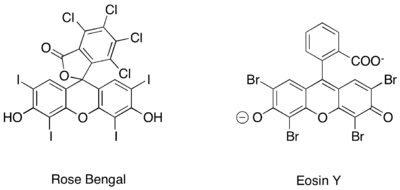
Podobné fotoredoxní tvorby iminiových iontů iminium bylo také dosaženo při použití zcela organických fotoredoxních katalyzátorů, jako jsou rose bengal a eosin Y.[14][15][16]
V asymetrické variantě této reakce se používají ekvivalenty acylových nukleofilů vytvářené za katalýzy N-heterocyklickými karbeny.[17]
Při tomto postupu se obchází problém slabé enantioindukce u chirálních fotoredoxních katalyzátorů přesunutím zdroje enantioselektivity na N-heterocyklický karben.
Oxidativní tvorba oxokarbeniových iontů
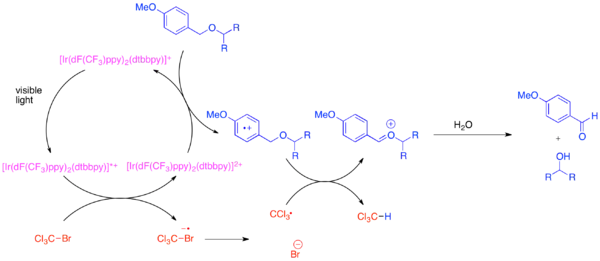
Rozvoj ortogonálních chránicích skupin v organické syntéze byl obtížný, protože tyto skupiny mohou rozlišovat mezi každými běžnými funkčními skupinami, jako jsou hydroxyly. Častou chránicí skupinou pro hydroxyly je para-methoxybenzylether (PMB ether). Tato skupina je chemicky podobná na elektrony méně bohatému benzyletheru. Odštěpení PMB etherové skupiny se obvykle provádí za přítomnosti benzyletheru pomocí silných stechiometrických oxidačních činidel, jako jsou 2,3-dichlor-5,6-dikyano-1,4-benzochinon (DDQ) nebo dusičnan amonno-ceričitý (CAN). PMB ethery jsou k oxidacím náchylnější než benzylethery, protože disponují větší elektronovou hustotou. Selektivní odštěpení PMB etherů lze dosáhnout s využitím bis-(2-(2',4'-difluorfenyl)-5-trifluormethylpyridin)-(4,4'-di-terc-butylbipyridin) hexafluorofosforečnqnu iriditého (Ir[dF(CF3)ppy]2(dtbbpy)PF6) a mírného stechiometrického oxidantu, například bromtrichlormethanu (BrCCl3).[18]
Fotoexcitovaný katalyzátor obsahující iridium je dostatečně silný na to, aby vyvolával fragmentaci bromtrichlormethanu na trichlormethylové radikály, bromidové anionty a iridičitý komplex. Díky fluorovaným ligandům je komplex dostatečně silným oxidačním činidlem, aby přijal elektrony z arenu s vysokou elektronovou hustotou, jako je PMB ether. Po zoxidování aren společně s trichlormethylovým radikálem vstupuje do přesunu vodíkového atomu, kde vytvoří chloroform a oxokarbeniový ion, který se následně hydrolyzuje za vzniku hydroxidu. Bylo prokázáno, že tato reakce je ortogonální vůči mnoha běžným chránicím skupinám, pokud se přidá zásada neutralizující vytvořený HBr.
Cykloadice
Cykloadice a ostatní pericyklické reakce mají velké využití v organické syntéze, protože je lze použít k rychlé tvorbě složitých struktur, částečně také díky možnosti vytvoření několika stereocenter ovladatelným způsobem. Podle Woodwardových–Hoffmannových pravidel orbitalové symetrie či jiných modelů, jako jsou teorie hraničních molekulových orbitalů (FMO) a Dewarův-Zimmermannův model, však mohou za tepelných podmínek probíhat pouze některé cykloadice. Cykloadice, které nejsou tepelně povolené, například [2+2] cykloadice, lze provést s využitím fotochemické aktivace. Za nepřítomnosti katalyzátoru tyto aktivace vyžadují použití velkých energií ultrafialového záření. Rovněž použit kovové katalyzátory, jako kobalt a měď, mohou katalyzovat tepelně nevýhodné [2+2] cykloadice přenosem jednoho elektronu.
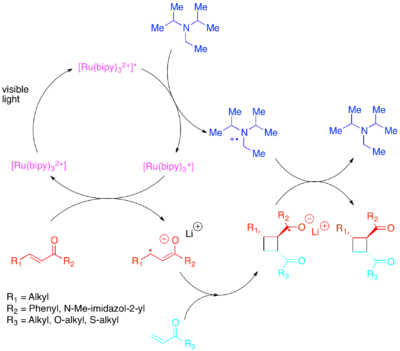
Potřebné změny v obsazení orbitalů lze dosáhnout přesunem elektronu pomocí fotokatalyzátoru citlivého na méně energetické viditelné světlo.[19][20][21][22][23]
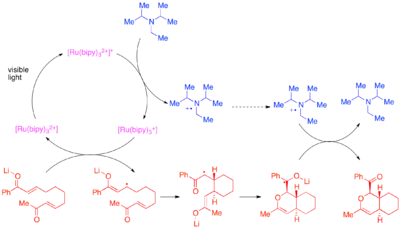
Tehshik P. Yoon provedl účinné vnitro- i mezimolekulové [2+2] cykloadice u aktivovaných alkenů; konkrétně šlo o enony a styreny. Enony, které mají nízkou elektronovou hustotu, reagují mechanismem zahrnujícím radikálové anionty, přičemž se jako zdroj elektronů používá diisopropylethylamin. Jako vhodný katalyzátor se ukázal komplex [Ru(bipy)3]2+. Bylo zjištěno, že aniontová povaha cyklizace je důležitá, jelikož by při provádění reakce s kyselinovým protiiontem místo lithného vedlo k převaze jiných než cykloadičních mechanismů.[24]
Jiný cyklizační postup lze použít u chalkonoidů se samaritým protiiontem.[25]
Naopak styreny, mající vysokou elektronovou hustotu, reagují skrze radikálové kationty, přičemž je možné k přenosu elektronů použít paraquat nebo molekulární kyslík. Zatímco [Ru(bipy)3]2+ je dobrým katalyzátorem vnitromolekulárních cyklizací s paraquatem, tak není vhodný u molekulárního kyslíku ani pro mezimolekulární cyklizace. U mezimolekulárních cyklizací, jak zjistili Yoon et al., tvoří silněji oxidující fotokatalyzátor [Ru(bpm)3]2+ a molekulový kyslík vhodnější katalytický systém pro tvorbu radikálových kationtů nutných k průběhu cykloadice. [Ru(bpz)3]2+, který je ještě silnějším oxidantem, není vhodný, protože i když může katalyzovat žádané [2+2] cykloadice, tak také může oxidovat cykloadukt a katalyzovat zpětnou [2+2] reakci. Toto srovnání ukazuje důležitost vhodné úpravy redoxních vlastností fotokatalyzátorů vůči reakčnímu systému a také využitelnost polypyridylových sloučenin jako ligandů, které lze lehce upravovat k zajištění vhodných redoxních vlastností jejich komplexů.
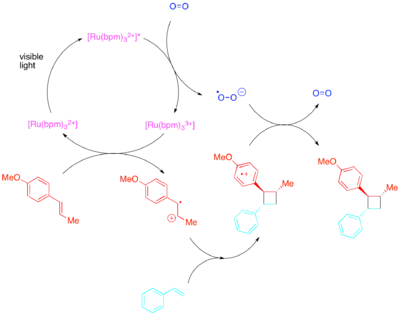
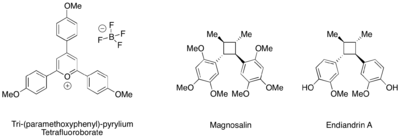
Výtěžnost fotoredoxně katalyzovaných [2+2] cykloadicí lze také vylepšit trifenylpyryliovými katalyzátory.[26]
Kromě tepelně nevýhodných [2+2] cykloadicí lze fotoredoxní katalýzu využít i u [4+2] cyklizací (Dielsových–Alderových reakcí). Bis-enony, podobné substrátům používaným při [2+2] fotoredoxních cyklizacích, ovšem s delšími můstky propojujícími dvě enonové skupiny, se mohou zapojit do vnitromolekulárních radikálově aniontových hetero-Dielsových–Alderových reakcí snadněji než do [2+2] cykloadicí.[27]
Podobně se styreny mohou účastnit vnitromolekulárních i mezimolekulárních Dielsových–Alderových reakcí probíhajících přes radikálové kationty.[28][29]
[Ru(bipy)3]2+ je vhodným katalyzátorem mezimolekulárních, ovšem nikoliv vnitromolekulárních, Dielsových–Alderových cyklizací. Tyto fotoredoxně katalyzované Dielsovy–Alderovy reakce dovolují uskutečnění cykloadičních reakcí dvou elektronově nesladěných substrátů. Obvyklé elektronové požadavky na Dielsovy–Alderovy reakce představují na elektrony bohatý dien, který reaguje s elektronově chudým alkenem (také se používá označení „dienofil“), zatímco elektronově inverzní Dielsovy–Alderovy reakce probíhají mezi na elektrony chudým dienem a dienofilem s vysokou elektronovou hustotou. Fotoredoxní varianta však probíhá jiným mechanismem než tepelná, a tak cykloadiční reakce na elektrony bohatého dienu s chudým dienofilem otevírá cestu k novým druhům Dielsových–Alderových produktů.
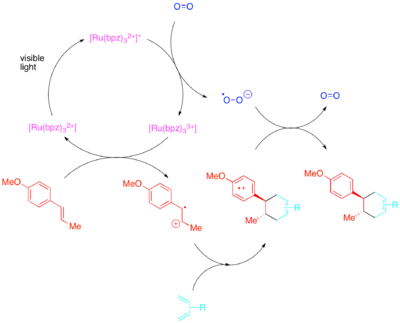
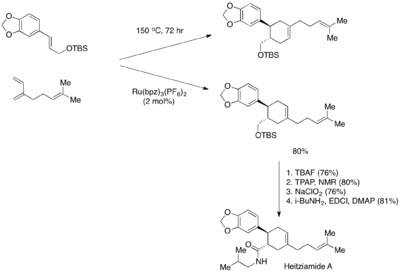
Syntetický význam Yoonovy fotoredoxně katalyzované Dielsovy–Alderovy reakce styrenu lze předvést na totální syntéze heitziamidu A.[28] Tato syntéza ukazuje, že tepelná Dielsova–Alderova reakce upřednostňuje nechtěný regioizomer, zatímco fotoredoxně katalyzovaná varianta má výnosnost požadovaného regioizomeru podstatně vyšší.
Fotoredoxní organokatalýza
Organokatalýza se zabývá katalytickým využitím malých organických molekul jako katalyzátorů, zvláště k enantioselektivní syntéze chirálních sloučenin. Jedním z postupů je použití chirálních sekundárních aminů k aktivaci karbonylových sloučenin. V tomto případě amin kondenzuje s karbonylovou sloučeninou a vytvoří nukleofilní enamin. Konkrétní izomer chirálního aminu vznikne díky tomu, že je strana molekuly enaminu je stericky stíněná a tak se pouze druhá strana může účastnit reakce. I přes využitelnost tohoto postupu ke katalyzování enantioselektivní funkcionalizace karbonylových sloučenin, takto nelze provést některé důležité přeměny, jako jsou katalytická enantioselektivní α-alkylace aldehydů. Spojením organokatalýzy a fotoredoxních metod je možné= tento nedostatek překonat.[30]
Při α-alkylaci aldehydů [Ru(bipy)3]2+ redukčně fragmentuje aktivovaný alkylhalogenid, například bromomalonát či fenacylbromid, který se následně aduje na katalyticky tvořený enamin, přičemž reakce probíhá enantioselektivně. Oxidovaný fotokatalyzátor následně oxidativně odštěpí vzniklý α-aminový radikál za vzniku iminiového iontu, jenž je hydrolyzován na funktionalizovanou karbonylovou sloučeninu. Tato fotoredoxní přeměna je mechanisticky odlišná od jiného organokatalytického radikálového procesu nazývaného katalýza jednonásobně obsazenými molekulovými orbitaly (SOMO). Při SOMO katalýze se používá nadstechiometrické množství dusičnanu amonno-ceričitého k oxidaci katalyticky vzniklého enaminu na radikálový kation, jenž je poté adován na vhodný substrát, například allylsilany. Tento druh mechanismu není možný u fotokatalytických alkylací, protože vznikající enaminové radikálové kationty se cyklizují na cyklopropanové radikály, které jsou vůči fotoredoxní katalýze nereaktivní.
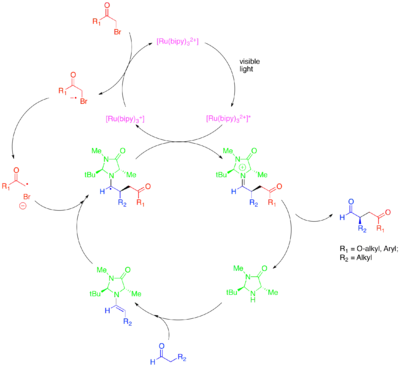
K této skupině reakcí patří také alkylace jinými druhy aktivovaných halogenalkanů. Použití fotokatalyzátoru Ir(dtbbpy)(ppy)2+ dovoluje enantioselektivní α-trifluormethylaci aldehydů a při použití Ir(ppy)3 je možné provést enantioselektivní reakce aldehydů s benzylbromidy chudými na elektrony.[31][32]
Zeitler et al. také prozkoumali spojení fotoredoxních a organokatalytických metod za účelem dosažení enantioselektivní alkylace aldehydů.[33] Stejné chirální imidazolidinonové organokatalyzátory byly využity k přípravě enaminu a zavedení chirality. Organický fotoredoxní katalyzátor eosin Y byl však používán častěji než komplexy ruthenia či iridia.
Přímá β-arylace nasycených aldehydů a ketonů může být zefektivněna pomocí fotoredoxní a organické katalýzy.[34] Předchozí postup spočíval v přímé β-funkcionalizaci nasycených karbonylových sloučenin jednohrnkově?? skládající se ze dvou kroků, obou katalyzovaných sekundárním aminem: stechiometrické redukce aldehydu pomocí IBX a reakce s aktivovaným alkylovým nukleofilem na pozici beta za vzniku výsledného enalu.[35] Tento proces, podobně jako i jiné fotoredoxní děje, probíhá radikálovým mechanismem, a je omezen na adice silně elektrofilních arenů na pozice beta. Výrazné omezení spektra použitelných arenů plyne především z nutnosti tvorby arenového radikálového aniontu dostatečně stabilního na to, aby nereagoval přímo s enaminem nebo enaminovým radikálovým kationtem. V navrženém mechanismu aktivovaný fotoredoxní katalyzátor oxidativně reaguje s arenem chudým na elektrony, jako je například 1,4-dikyanobenzen. Fotokatalyzátor následně oxiduje enamin, průběžně vytvořený kondenzací aldehydu se sekundárním aminem sloužícím jako kokatalyzátor (zde může jít například o isopropylbenzylamin). Vzniklý enaminový radikálový kation obvykle reaguje jako systém obsahující 3 π elektrony, ovšem z důvodu stability příslušných radikálů vzniká deprotonací na β-methylenové pozici systém 5 π elektronů se silně radikálovými vlastnostmi na nově zpřístupněném β-uhlíku. I když je při této reakci nutné použití sekundárního aminu jako organokatalyzátoru, protože se tím dosahuje tvorby enaminu, jenž je poté oxidován, tak není známa žádná její enantioselektivní varianta.
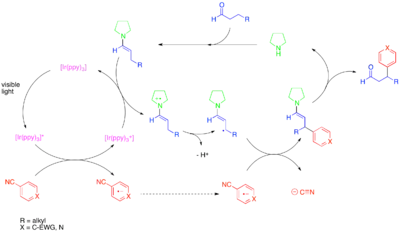
Rozvoj přímých β-arylací aldehydů vedl k vývoji podobných β-funkcionalizací cyklických ketonů; β-arylace cyklických ketonů přitom probíhají za podobných podmínek, ovšem za použití azepanu jakožto sekundárního aminového kokatalyzátoru. U cyklických ketonů lze uskutečnit fotokatalytické „homoaldolové“ reakce, a to díky umožnění párování na pozici beta vůči ipso uhlíku arylketonu, například benzofenonu či acetofenonu.[36]
Kromě azepanového kokatalyzátoru je také potřeba silněji redukující fotoredoxní katalyzátor Ir(ppy)3 a přidání hexafluoroarsenidu lithného (LiAsF6) do reakční směsi, které spouští jednoelektronovou redukci arylketonu.
Adice na alkeny
Využití fotoredoxní katalýzy k tvorbě reaktivních radikálů s heteroatomovými centry začalo být zkoumáno v 90. letech 20. století.[37]
[Ru(bipy)3]2+ katalyzuje fragmentaci tosylfenylselenidu na fenylselenolátový anion a tosylový radikál a tím tedy radikálové prodlužování řetězce, které umožní adici tosylového a fenylselenového radikálu na dvojnou vazbu na elektrony bohatých alkylvinyletherů. Protože je fenylselenolátový anion is snadno oxidovatelný na difenyldiselenid, tak malé množství pozorovaných difenyldiselenidových iontů bylo přičteno tomu, že fotoredoxně katalyzovaná fragmentace tosylfenylselenidu má význam jen v úvodním kroku, a většinu reaktivity způsobuje radikálový řetězový proces.
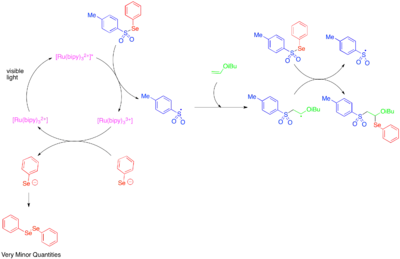
Heteroaromatické adice na alkeny zahrnují vícesložkové oxy- a aminotrifluormethylační reakce.[38][39]
Při těchto reakcích se používá Umemotovo činidlo, což je sulfoniová sůl sloužící jako elektrofilní zdroj trifluormethylových skupin; předpokládá se, že reaguje jednoelektronovým mechanismem. Jednoelektronovou redukcí Umemotova činidla vznikají trifluormethylové radikály, které se navazují na reaktivní alken. Následně se jednoelektronovou oxidací vzniklého alkylového radikálu utvoří kation, který může být zachycen vodou, alkoholem nebo nitrilem. Dosažení vysoké regioselektivity bylo popsáno hlavně u styrenů, které jsou odolné vůči tvorbě benzylových radikálových meziproduktů.
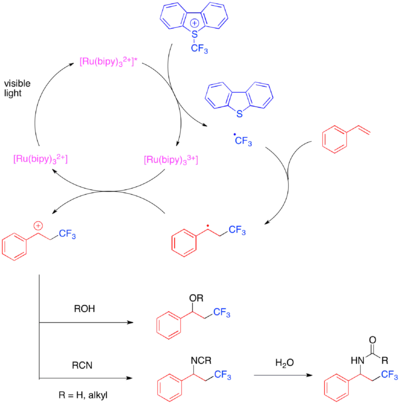
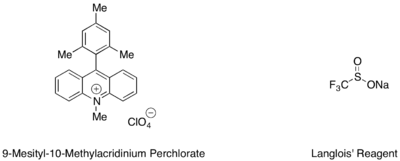
Výtěžnost hydrotrifluormethylací styrenů a alifatických alkenů lze zlepšit přidáním mesitylakridiniového katalyzátoru a Langloisova činidla jako zdroje CF3 radikálů.[40]
Jako nejvhodnější zdroje vodíkových radikálů, umožňujících zakončení katalytického cyklu, se ukázaly směsi trifluorethanolu s podstechiometrickými množstvími aromatického thiolu, například methylthiosalicylátu.
Vnitromolekulární hydroetherifikace a hydroaminace probíhají proti Markovnikovovu pravidlu.[41][42]
Jeden mechanismus zahrnuje jednoelektronovou oxidaci alkenu, zachycení radikálového kationtu hydroxylovou nebo aminovou funkční skupinou a přesun vodíkového atomu z reaktivního donoru na alkylový radikál. Rozšíření této reaktivity na mezimolekulární systémy způsobuje i) nové způsoby syntézy tetrahydrofuranů reakcemi allylalkoholů s alkeny a ii) adici karboxylových kyselin na alkeny probíhající proti Markovnikovovu pravidlu.[43][44]
Reference
V tomto článku byl použit překlad textu z článku Photoredox catalysis na anglické Wikipedii.
- Nathan A. Romero; David A. Nicewicz. Organic Photoredox Catalysis. Chemical Reviews. 2016-06-10, s. 10075–10166. DOI 10.1021/acs.chemrev.6b00057. PMID 27285582.
- Wayne E. Jones; Marye Anne Fox. Determination of Excited-State Redox Potentials by Phase-Modulated Voltammetry. The Journal of Physical Chemistry. 1994, s. 5095–5099. DOI 10.1021/j100070a025.
- Electrochemical Series of Photocatalysts and Common Organic Compounds [online]. Merck [cit. 2019-04-15]. Dostupné v archivu pořízeném dne 2015-12-16.
- Joseph W. Tucker; Corey R. J. Stephenson. Shining Light on Photoredox Catalysis: Theory and Synthetic Applications. The Journal of Organic Chemistry. 2012, s. 1617–1622. DOI 10.1021/jo202538x. PMID 22283525.
- Jagan M. R. Narayanam; Joseph W. Tucker; Corey R. J. Stephenson. Electron-Transfer Photoredox Catalysis: Development of a Tin-Free Reductive Dehalogenation Procedure. Journal of the American Chemical Society. 2009-06-05, s. 8756–8757. DOI 10.1021/ja9033582. PMID 19552447.
- John D. Nguyen; Erica M. D'Amato; Jagan M. R. Narayanam; Corey R. J. Stephenson. Engaging unactivated alkyl, alkenyl and aryl iodides in visible-light-mediated free radical reactions. Nature Chemistry. 2012, s. 854–859. DOI 10.1038/nchem.1452. PMID 23001000.
- Laura Furst; Jagan M. R. Narayanam; Corey R. J. Stephenson. Total Synthesis of (+)-Gliocladin C Enabled by Visible-Light Photoredox Catalysis. Angewandte Chemie International Edition. 2011-10-04, s. 9655–9659. DOI 10.1002/anie.201103145. PMID 21751318.
- Allison G. Condie; José C. González-Gómez; Corey R. J. Stephenson. Visible-Light Photoredox Catalysis: Aza-Henry Reactions via C−H Functionalization. Journal of the American Chemical Society. 2010-02-10, s. 1464–1465. DOI 10.1021/ja909145y. PMID 20070079.
- Magnus Rueping; Shaoqun Zhu; René M. Koenigs. Visible-light photoredox catalyzed oxidative Strecker reaction. Chemical Communications. 2011, s. 12709–12711. DOI 10.1039/C1CC15643H. PMID 22041859.
- Guolei Zhao; Chao Yang; Lin Guo; Hongnan Sun; Chao Chen; Wujiong Xia. Visible light-induced oxidative coupling reaction: easy access to Mannich-type products. Chemical Communications. 2012, s. 2337–2339. DOI 10.1039/C2CC17130A. PMID 22252544.
- Magnus Rueping; Shaoqun Zhu; René M. Koenigs. Photoredox catalyzed C–P bond forming reactions—visible light mediated oxidative phosphonylations of amines. Chemical Communications. 2011, s. 8679–8681. DOI 10.1039/C1CC12907D. PMID 21720622.
- David B. Freeman; Laura Furst; Allison G. Condie; Corey R. J. Stephenson. Functionally Diverse Nucleophilic Trapping of Iminium Intermediates Generated Utilizing Visible Light. Organic Letters. 2012, s. 94–97. DOI 10.1021/ol202883v. PMID 22148974.
- Magnus Rueping; René M. Koenigs; Konstantin Poscharny; David C. Fabry; Daniele Leonori; Carlos Vila. Dual Catalysis: Combination of Photocatalytic Aerobic Oxidation and Metal Catalyzed Alkynylation Reactions-C≡C Bond Formation Using Visible Light. Chemistry: A European Journal. 2012-04-23, s. 5170–5174. DOI 10.1002/chem.201200050.
- Yuanhang Pan; Shuai Wang; Choon Wee Kee; Emilie Dubuisson; Yuanyong Yang; Kian Ping Loh; Choon-Hong Tan. Graphene oxide and Rose Bengal: oxidative C–H functionalisation of tertiary amines using visible light. Green Chemistry. 2011, s. 3341. DOI 10.1039/C1GC15865A.
- Weijun Fu; Wenbo Guo; Guanglong Zou; Chen Xu. Selective trifluoromethylation and alkynylation of tetrahydroisoquinolines using visible light irradiation by Rose Bengal. Journal of Fluorine Chemistry. 2012, s. 88–94. DOI 10.1016/j.jfluchem.2012.05.009.
- Durga Prasad Hari; Burkhard König. Eosin Y Catalyzed Visible Light Oxidative C–C and C–P bond Formation. Organic Letters. 2011-08-05, s. 3852–3855. DOI 10.1021/ol201376v. PMID 21744842.
- Daniel A. DiRocco; Tomislav Rovis. Catalytic Asymmetric α-Acylation of Tertiary Amines Mediated by a Dual Catalysis Mode: N-Heterocyclic Carbene and Photoredox Catalysis. Journal of the American Chemical Society. 2012-05-16, s. 8094–8097. DOI 10.1021/ja3030164. PMID 22548244.
- Joseph W. Tucker; Jagan M. R. Narayanam; Pinkey S. Shah; Corey R. J. Stephenson. Oxidative photoredox catalysis: mild and selective deprotection of PMB ethers mediated by visible light. Chemical Communications. 2011, s. 5040–5042. DOI 10.1039/c1cc10827a. PMID 21431223.
- Michael A. Ischay; Mary E. Anzovino; Juana Du; Tehshik P. Yoon. Efficient Visible Light Photocatalysis of [2+2] Enone Cycloadditions. Journal of the American Chemical Society. 2008, s. 12886–12887. DOI 10.1021/ja805387f. PMID 18767798.
- Juana Du; Tehshik P. Yoon. Crossed Intermolecular [2+2] Cycloadditions of Acyclic Enones via Visible Light Photocatalysis. Journal of the American Chemical Society. 2009-10-21, s. 14604–14605. DOI 10.1021/ja903732v. PMID 19473018.
- Michael A. Ischay; Zhan Lu; Tehshik P. Yoon. [2+2] Cycloadditions by Oxidative Visible Light Photocatalysis. Journal of the American Chemical Society. 2010-06-30, s. 8572–8574. DOI 10.1021/ja103934y. PMID 20527886.
- Elizabeth L. Tyson; Elliot P. Farney; Tehshik P. Yoon. Photocatalytic [2 + 2] Cycloadditions of Enones with Cleavable Redox Auxiliaries. Organic Letters. 2012-02-17, s. 1110–1113. DOI 10.1021/ol3000298. PMID 22320352.
- Michael A. Ischay; Michael S. Ament; Tehshik P. Yoon. Crossed intermolecular [2 + 2] cycloaddition of styrenes by visible light photocatalysis. Chemical Science. 2012, s. 2807–2811. DOI 10.1039/c2sc20658g. PMID 22984640.
- Juana Du; Laura Ruiz Espelt; Ilia A. Guzei; Tehshik P. Yoon. Photocatalytic reductive cyclizations of enones: Divergent reactivity of photogenerated radical and radical anion intermediates. Chemical Science. 2011, s. 2115–2119. DOI 10.1039/c1sc00357g. PMID 22121471.
- Guolei Zhao; Chao Yang; Lin Guo; Hongnan Sun; Run Lin; Wujiong Xia. Reactivity Insight into Reductive Coupling and Aldol Cyclization of Chalcones by Visible Light Photocatalysis. The Journal of Organic Chemistry. 2012-07-20, s. 6302–6306. DOI 10.1021/jo300796j. PMID 22731518.
- Michelle Riener; David A. Nicewicz. Synthesis of cyclobutane lignans via an organic single electron oxidant–electron relay system. Chemical Science. 2013, s. 2625. DOI 10.1039/c3sc50643f. PMID 24349680.
- Anna E. Hurtley; Megan A. Cismesia; Michael A. Ischay; Tehshik P. Yoon. Visible light photocatalysis of radical anion hetero-Diels–Alder cycloadditions. Tetrahedron. 2011, s. 4442–4448. DOI 10.1016/j.tet.2011.02.066. PMID 21666769.
- Shishi Lin; Michael A. Ischay; Charles G. Fry; Tehshik P. Yoon. Radical Cation Diels–Alder Cycloadditions by Visible Light Photocatalysis. Journal of the American Chemical Society. 2011-12-07, s. 19350–19353. DOI 10.1021/ja2093579. PMID 22032252.
- Shishi Lin; Christian E. Padilla; Michael A. Ischay; Tehshik P. Yoon. Visible light photocatalysis of intramolecular radical cation Diels–Alder cycloadditions. Tetrahedron Letters. 2012, s. 3073–3076. DOI 10.1016/j.tetlet.2012.04.021. PMID 22711942.
- D. A. Nicewicz; D. W. C. MacMillan. Merging Photoredox Catalysis with Organocatalysis: The Direct Asymmetric Alkylation of Aldehydes. Science. 2008-10-03, s. 77–80. DOI 10.1126/science.1161976. PMID 18772399.
- David A. Nagib; Mark E. Scott; David W. C. MacMillan. Enantioselective α-Trifluoromethylation of Aldehydes via Photoredox Organocatalysis. Journal of the American Chemical Society. 2009-08-12, s. 10875–10877. DOI 10.1021/ja9053338. PMID 19722670.
- Hui-Wen Shih; Mark N. Vander Wal; Rebecca L. Grange; David W. C. MacMillan. Enantioselective α-Benzylation of Aldehydes via Photoredox Organocatalysis. Journal of the American Chemical Society. 2010-10-06, s. 13600–13603. DOI 10.1021/ja106593m. PMID 20831195.
- Matthias Neumann; Stefan Füldner; Burkhard König; Kirsten Zeitler. Metal-Free, Cooperative Asymmetric Organophotoredox Catalysis with Visible Light. Angewandte Chemie International Edition. 2011-01-24, s. 951–954. DOI 10.1002/anie.201002992. PMID 20878819.
- M. T. Pirnot; D. A. Rankic; D. B. C. Martin; David W. C. MacMillan. Photoredox Activation for the Direct-Arylation of Ketones and Aldehydes. Science. 2013-03-28, s. 1593–1596. DOI 10.1126/science.1232993. PMID 23539600.
- Shi-Lei Zhang; He-Xin Xie; Jin Zhu; Hao Li; Xin-Shuai Zhang; Jian Li; Wei Wang. Organocatalytic enantioselective β-functionalization of aldehydes by oxidation of enamines and their application in cascade reactions. Nature Communications. 2011-03-01, s. 211. DOI 10.1038/ncomms1214. PMID 21364550.
- Filip R. Petronijević; Manuel Nappi; David W. C. MacMillan. Direct β-Functionalization of Cyclic Ketones with Aryl Ketones via the Merger of Photoredox and Organocatalysis. Journal of the American Chemical Society. 2013-01-22. DOI 10.1021/ja410478a. PMID 24237366.
- Derek H. R. Barton; Maria A. Csiba; Joseph Cs. Jaszberenyi. Ru(bpy)32+-mediated addition of Se-phenyl p-tolueneselenosulfonate to electron rich olefins. Tetrahedron Letters. 1994, s. 2869–2872. DOI 110.1016/S0040-4039(00)76646-9.
- Yusuke Yasu; Takashi Koike; Munetaka Akita. Three-component Oxytrifluoromethylation of Alkenes: Highly Efficient and Regioselective Difunctionalization of C=C Bonds Mediated by Photoredox Catalysts. Angewandte Chemie International Edition. 2012-09-17, s. 9567–9571. DOI 10.1002/anie.201205071. PMID 22936394.
- Yusuke Yasu; Takashi Koike; Munetaka Akita. Intermolecular Aminotrifluoromethylation of Alkenes by Visible-Light-Driven Photoredox Catalysis. Organic Letters. 2013-05-03, s. 2136–2139. DOI 10.1021/ol4006272. PMID 23600821.
- Dale J. Wilger; Nathan J. Gesmundo; David A. Nicewicz. Catalytic hydrotrifluoromethylation of styrenes and unactivated aliphatic alkenes via an organic photoredox system. Chemical Science. 2013, s. 3160. DOI 10.1039/c3sc51209f.
- David S. Hamilton; David A. Nicewicz. Direct Catalytic Anti-Markovnikov Hydroetherification of Alkenols. Journal of the American Chemical Society. 2012, s. 18577–18580. DOI 10.1021/ja309635w. PMID 23113557.
- M. Tien; David A. Nicewicz. Anti-Markovnikov Hydroamination of Alkenes Catalyzed by an Organic Photoredox System. Journal of the American Chemical Society. 2013-07-03, s. 9588–9591. DOI 10.1021/ja4031616. PMID 23768239.
- Jean-Marc M. Grandjean; David A. Nicewicz. Synthesis of Highly Substituted Tetrahydrofurans by Catalytic Polar-Radical-Crossover Cycloadditions of Alkenes and Alkenols. Angewandte Chemie International Edition. 2013-04-02, s. 3967–3971. DOI 10.1002/anie.201210111. PMID 23440762.
- Andrew J. Perkowski; David A. Nicewicz. Direct Catalytic Anti-Markovnikov Addition of Carboxylic Acids to Alkenes. Journal of the American Chemical Society. 2013-07-17, s. 10334–10337. DOI 10.1021/ja4057294. PMID 23808532.