Sharplessova asymetrická dihydroxylace
Sharplessova asymetrická dihydroxylace (také se používá název Sharplessova bishydroxylace) je chemická reakce alkenu s oxidem osmičelým za přítomnosti chirálního chininového ligandu, přičemž produktem je vicinální diol.
Tuto reakci je možné provést u alkenů s téměř jakýmikoliv substituenty, často vysoce ernantioselektivně. Asymetrické dihydroxylace bývají také značně regioselektivní, protože se tvoří převážně produkty reakcí na dvojných vazbách s největší elektronovou hustotou.[1][2][3]
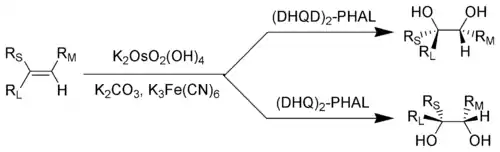
RL = největší substituent; RM = střední substituent; RS = nejmenší substituent
Reakce se často provádí za přítomnosti katalytického množství oxidu osmičelého, který se po reakci obnovuje například hexakyanoželezitanem draselným[4][5] nebo N-methylmorfolin N-oxidem.[6][7]
Tímto se výrazně snižuje potřebné množství toxického a nákladného oxidu osmičelého. Čtveřice reaktantů pro tuto reakci je komerčně dostupná jako směs.[8]
Některé chirální dioly mají využití v organické syntéze. Zavádění chirality do nechirálních molekul prostřednictvím chirálních katalyzátorů má velký význam.
Tuto reakci rozvinul Karl Barry Sharpless na základě již známé Upjohnovy dihydroxylace. V roce 2001 za to obdržel Nobelovu cenu za chemii.
Historie
Dihydroxylace alkenů osmičelým (OsO4) je poměrně starým, ovšem užitečným postupem funkcionalizace alkenů. Protože je však OsO4 drahý a vysoce toxický, tak se objevily snahy o vývoj katalytických variant. Jako stechiometrická oxidační činidla se používají například chlorečnan draselný, peroxid vodíku, N-methylmorfolin N-oxid, terc-butylhydroperoxid a hexakyanoželezitan draselný (K3Fe(CN)6). Karl Barry Sharpless jako první provedl všeobecnou a použitelnou enantioselektivní dihydroxylaci alkenů. Malá množství OsO4 se smíchají se stechiometrickým množstvím hexakyanoželezitanu za přítomnosti chirálních dusíkatých ligandů, čímž se vytvoří okolo oxidačního činidla asymetrické prostředí.
Mechanismus
Sharplessova dihydroxylace začíná tvorbou komplexu oxidu osmičelého a ligandu (2). [3+2]-cykloadicí s alkenem (3) se vytvoří cyklický meziprodukt 4.[9][10] Zásaditou hydrolýzou se uvolní diol (5) a redukovaný osman (6). Tuto část katalytického cyklu urychluje methansulfonamid (CH3SO2NH2), a tak se často přidává do reakční směsi za účelem dosažení dobrého průběhu reakce nekoncových alkenů i při 0 °C.[8] Nakonec stechiometrický oxidant obnoví komplex oxidu osmičelého a ligandu (2).
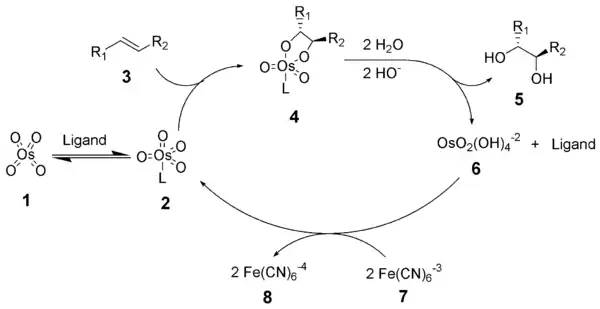
Mechanismus Sharplessovy dihydroxylace byl podrobně prozkoumán a byl nalezen možný druhý katalytický cyklus (viz níže).[11][12]
Pokud je osmylátový esterový meziprodukt zoxidován před tím, než se stihne disociovat, tak se vytvoří komplex OsVIII-diol, jenž může dihydroxylovat další alken.[13] Dihydroxylace touto vedlejší drahou se obvykle vyznačují nižšími enantioselektivitami než u hlavní dráhy; lze ji potlačit zvýšením molární koncentrace ligandu.
Níže je zobrazen vedlejší katalytický cyklus:
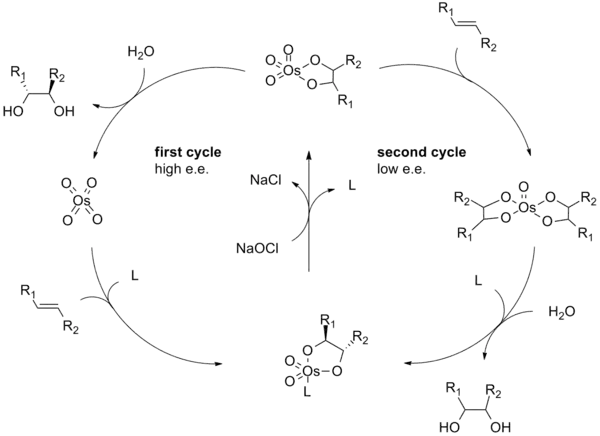
[2+2]/[3+2] mechanismus
Sharpless původně navrhl, že tato reakce probíhá jako [2+2] cykloadice OsO4 na alken za vzniku osmaoxetanového meziproduktu.[14] Tento meziprodukt měl následně projít 1,1-migrační insercí za tvorby osmylátového esteru, z něhož po hydrolýze vznikl diol. V roce 1989 E. J. Corey popsal mírně odlišnou variantu reakce a prohlásil, že nejpravděpodobnější je [3+2] cykloadice OsO4 na alken za přímé tvorby osmylátového esteru.[15]
Coreyův výzkum byl založen na předchozích výpočetních studiích, ve kterých se ukázalo, že [3+2] mechanismus má nižší aktivační energii. Corey navíc zjistil, že sterické odpuzování v oktaedrickém meziproduktu způsobuje nevýhodnost [2+2] mechanismu.

Během následujících 10 let vydali Corey i Sharpless, několik prací, kde každý podporoval svou verzi mechanismu. I když tyto studie nemohly rozhodnout mezi oběma reakčními drahami, tak vyloučily možnost některých jiných průběhů, například Sharpless dokázal, že reakce probíhá postupným mechanismem.[16] Následně oba zjistili, že aktivní katalyzátor obsahuje chirální „vazebnou kapsu“ ve tvaru U.[17][18][19]
Corey také ukázal , že katalyzátor má vlastnosti odpovídající Michaelisovské–Mentenovské–Monodovské kinetice.[20]
V roce 1997 Sharpless vydal výsledky studie, které podpořily [2+2] cyklizaci oproti [3+2].[21]
Později ve stejném roce také vydal studii, která potvrzovala [3+2] mechanismus[10] a byl tak nucen ukončit desetiletý spor.
Struktura katalyzátoru
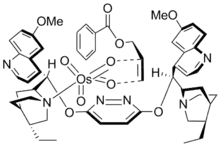
Pomocí krystalografie bylo zjištěno, že aktivní katalyzátor je pětikoordinovaná sloučenina osmia uvnitř vazebné kapsy ve tvaru U. Dusíkatý ligand udržuje OsO4 v chirálním prostředí a způsobuje, že jedna strana alkenu je stericky stíněná a druhá není.[20]
Katalytické systémy
Pro Sharplessovy asymetrické dihydroxylace bylo vyvinuto několik katalytických systémů. Níže je zobrazen přehled jejich složek:
- Katalytický oxidant: Touto složkou je pokaždé OsO4, i když lze pro koordinaci OsVIII a úpravě jeho elektronových vlastností přidat další látky. OsO4 se z bezpečnostních důvodů připravuje na místě z K2OsO2(OH)4 (sloučeniny OsVI).
- Chirální pomocník: Obvykle některý z cinchonových alkaloidů.
- Stechiometrický oxidant:
- Jako jedny z prvních stechiometrických oxidantů se používaly peroxidy, mají však nevýhodu ohledně nízké chemoselektivity.[13]
- N-oxidy trialkylamonných sloučenin , například NMO a trimethylamin-N-oxid.[13]
- Nejčastěji používaným stechiometrickým oxidačním činidlem je hexakyanoželezitan draselný (K3Fe(CN)6), ten je také složkou komerčně dostupných směsí.
- Přídavné látky:
- Kyselina citronová: Oxid osmičelý je elektrofilní oxidant a tak s alkeny s nízkou elektronovou hustotou reaguje pomalu. Rychlost oxidace takových alkenů lze ovšem urychlit mírným snížením pH reakční směsi;[13] vyšší pH však může urychlit oxidaci vnitřních alkenů a také zlepšit enantiomerní přebytky u oxidací koncových alkenů.[13]
Regioselektivita
U obecných Sharplessových asymetrických dihydroxylací se snadněji oxidují alkeny bohatší na elektrony.[22]
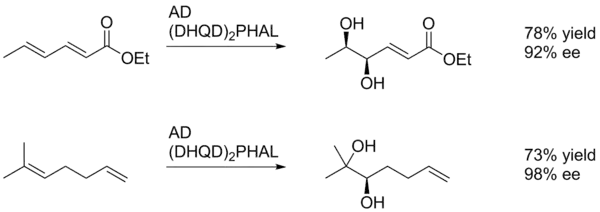
V tomto případě diol vzniká na alkenové skupině nejbližší k p-methoxybenzoylu, výtěžnost je ale nízká. Selektivitu vyvolávají příznivé interakce arylového jádra s aktivním místem katalyzátoru. Arylový substituent zde může působit jako řídicí skupina.
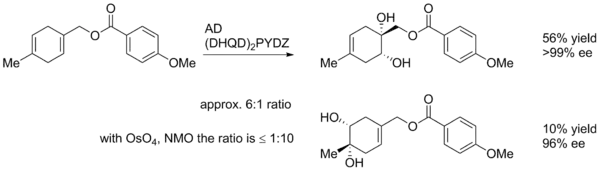
Stereoselektivita
Diastereoselektivitu Sharplessových asymetrických dihydroxylací lze nejlépe ovládat volbou ligandu (například mezi AD-mix-α a AD-mix-β), ovšem vliv mohou mít i jiné faktory, jako jsou již existující chiralita na substrátu či substituenty. V níže uvedeném příkladě je použita p-methoxybenzoylová skupina jako zdroj sterického napětí, které umožní katalyzátoru rozlišit dvě strany alkenu.[23]
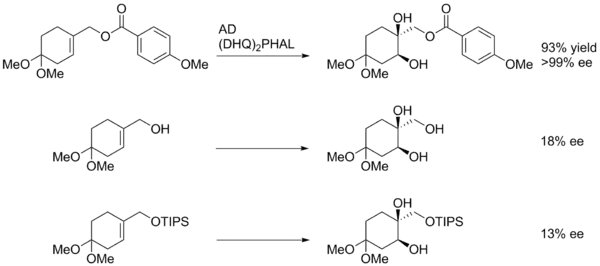
Dosažení vysoké diastereoselektivity bývá obtížné na cis-alkenech, kde jsou sterické podmínky na obou stranách alkenu podobné.
Odkazy
Externí odkazy
- E. N. Jacobsen; I. MARKO; W. S. MUNGALL; G. SCHROEDER; K. B. SHARPLESS. Asymmetric dihydroxylation via ligand-accelerated catalysis. Journal of the American Chemical Society. 1988, s. 1968–1970. DOI 10.1021/ja00214a053. (anglicky)
Související články
- Milasova hydroxylace
- Upjohnova dihydroxylace
- Sharplessova aminohydroxylace
Reference
V tomto článku byl použit překlad textu z článku Sharpless asymmetric dihydroxylation na anglické Wikipedii.
- Mark C. Noe; Michael A. Letavic; Sheri L. Snow. Asymmetric Dihydroxylation of Alkenes. Organic Reactions. 2005-12-15, s. 109–625. ISBN 0471264180. DOI 10.1002/0471264180.or066.02.
- H. C. Kolb; M. S. Van Nieuwenhze; K. B. Sharpless. Catalytic Asymmetric Dihydroxylation. Chemical Reviews. 1994, s. 2483–2547. ISBN 0471264180. DOI 10.1021/cr00032a009.
- GONZALEZ, Javier; AURIGEMMA, Christine; TRUESDALE, Larry. Synthesis of (+)-(1S,2R)- and (−)-(1R,2S)-trans-2-Phenylcyclohexanol Via Sharpless Asymmetric Dihydroxylation (AD). Org. Synth.. 2004, s. 93. Dostupné online. DOI 10.15227/orgsyn.079.0093. (anglicky)
- M. Minato; K. Yamamoto; J. Tsuji. Osmium tetraoxide catalyzed vicinal hydroxylation of higher olefins by using hexacyanoferrate(III) ion as a cooxidant. The Journal of Organic Chemistry. 1990, s. 766–768. DOI 10.1021/jo00289a066.
- OI, R.; SHARPLESS, K. B. 3-[(1S)-1,2-Dihydroxyethyl]-1,5-Dihydro-3H-2,4-Benzodioxepine. Org. Synth.. 1996, s. 1. Dostupné online. DOI 10.15227/orgsyn.073.0001. (anglicky); Coll. Vol.. S. 251. (anglicky)
- V. VanRheenen; R. C. Kelly; D., Y. Cha. An improved catalytic OsO4 oxidation of olefins to cis-1,2-glycols using tertiary amine oxides as the oxidant. Tetrahedron Letters. 1976, s. 1973–1976. DOI 10.1016/s0040-4039(00)78093-2.
- MCKEE, B. H.; GILHEANY, D. G.; SHARPLESS, K. B. (R,R)-1,2-Diphenyl-1,2-ethanediol (Stilbene diol). Org. Synth.. 1992, s. 47. Dostupné online. DOI 10.15227/orgsyn.070.0047. (anglicky); Coll. Vol.. S. 383. (anglicky)
- SHARPLESS, K. B.; AMBERG, Willi; BENNANI, Youssef L.; CRISPINO, Gerard A.; HARTUNG, Jens; JEONG, Kyu Sung; KWONG, Hoi Lun. The osmium-catalyzed asymmetric dihydroxylation: A new ligand class and a process improvement. The Journal of Organic Chemistry. 1992, s. 2768–2771. DOI 10.1021/jo00036a003. (anglicky)
- E. J. Corey; M. C. Noe; M. J. Grogan. Experimental test of the [3+2]- and [2+2]-cycloaddition pathways for the bis-cinchona alkaloid-OsO4 catalyzed dihydroxylation of olefins by means of kinetic isotope effects. Tetrahedron Letters. 1996, s. 4899–4902. DOI 10.1016/0040-4039(96)01005-2.
- A. J. DelMonte; J. Haller; K. N. Houk; K. B. Sharpless; D. A. Singleton; T. Strassner; A. A. Thomas. Experimental and Theoretical Kinetic Isotope Effects for Asymmetric Dihydroxylation. Evidence Supporting a Rate-Limiting "(3 + 2)" Cycloaddition. Journal of the American Chemical Society. 1997, s. 9907–9908. DOI 10.1016/0040-4039(96)01005-2.
- Y. Ogino; H. Chen; H.-L. Kwong; K. B. Sharpless. On the timing of hydrolysis / reoxidation in the osmium-catalyzed asymmetric dihydroxylation of olefins using potassium ferricyanide as the reoxidant. Tetrahedron Letters. 1991, s. 3965–3968. DOI 10.1016/0040-4039(91)80601-2.
- J. S. M. Wai; I. Marko; J. N. Svendsen; M. G. Finn; E. N. Jacobsen; K. B. Sharpless. A mechanistic insight leads to a greatly improved osmium-catalyzed asymmetric dihydroxylation process. Journal of the American Chemical Society. 1989, s. 1123. DOI 10.1021/ja00185a050.
- Sundermeier, U., Dobler, C., Beller, M. Recent developments in the osmium-catalyzed dihydroxylation of olefins. Modern Oxidation Methods. 2004 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. ISBN 3-527-30642-0
- Steven G. Hentges; K. Barry Sharpless. Asymmetric induction in the reaction of osmium tetroxide with olefins. Journal of the American Chemical Society. 1980, s. 4263. DOI 10.1021/ja00532a050.
- E. J. Corey; Jardine DaSilva; Virgil Scott; Po Wai Yuen; Richard D. Connell. Enantioselective vicinal hydroxylation of terminal and E-1,2-disubstituted olefins by a chiral complex of osmium tetroxide. An effective controller system and a rational mechanistic model. Journal of the American Chemical Society. 1989, s. 9243. DOI 10.1021/ja00532a050.
- Thomas, G.; Sharpless, K. B. ACIEE 1993, 32, 1329
- E. J. Corey; Mark C. Noe. Rigid and highly enantioselective catalyst for the dihydroxylation of olefins using osmium tetraoxide clarifies the origin of enantiospecificity. Journal of the American Chemical Society. 1993, s. 12579. DOI 10.1021/ja00079a045.
- H. C. Kolb; P. G. Anderson; K. B. Sharpless. Toward an Understanding of the High Enantioselectivity in the Osmium-Catalyzed Asymmetric Dihydroxylation (AD). 1. Kinetics. Journal of the American Chemical Society. 1994, s. 1278. DOI 10.1021/ja00083a014.
- E. J. Corey; Mark C. Noe; Sarshar Sepehr. X-ray crystallographic studies provide additional evidence that an enzyme-like binding pocket is crucial to the enantioselective dihydroxylation of olefins by OsO4-bis-cinchona alkaloid complexes. Tetrahedron Letters. 1994, s. 2861. DOI 10.1016/s0040-4039(00)76644-5.
- E. J. Corey; Mark C. Noe; Sarshar Sepehr. Kinetic Investigations Provide Additional Evidence That an Enzyme-like Binding Pocket Is Crucial for High Enantioselectivity in the Bis-Cinchona Alkaloid Catalyzed Asymmetric Dihydroxylation of Olefins. Journal of the American Chemical Society. 1996-01-17, s. 319. DOI 10.1021/ja952567z.
- SHARPLESS, K. B.; GYPSER, Andreas; HO, Pui Tong; KOLB, Hartmuth C.; KONDO, Teruyuki; KWONG, Hoi-Lun; MCGRATH, Dominic V. Toward an Understanding of the High Enantioselectivity in the Osmium-Catalyzed Asymmetric Dihydroxylation. 4. Electronic Effects in Amine-Accelerated Osmylations. Journal of the American Chemical Society. 1997, s. 1840. DOI 10.1021/ja961464t. (anglicky)
- D. Xu; G. A. Crispino; K. B. Sharpless. Selective asymmetric dihydroxylation (AD) of dienes. Journal of the American Chemical Society. 1992, s. 7570–7571. DOI 10.1021/ja00045a043.
- E. J. Corey; Angel Guzman-Perez; Mark C. Noe. The application of a mechanistic model leads to the extension of the Sharpless asymmetric dihydroxylation to allylic 4-methoxybenzoates and conformationally related amine and homoallylic alcohol derivatives. Journal of the American Chemical Society. 1995, s. 10805–10816. DOI 10.1021/ja00149a003.