Hydroaminace
Hydroaminace je organická reakce, při níž dochází k adici vazby N-H v molekule aminu na násobnou vazbu mezi uhlíkovými atomy.[1]
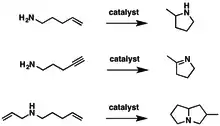
Hydroaminace může probíhat jako vnitromolekulární reakce (přitom vznikají heterocyklické sloučeniny) nebo mezimolekulárně (pak vznikne amin a nenasycená sloučenina). Stále se vyvíjejí nové katalyzátory hydroaminace, a to obzvláště v případě hydroaminace alkenů.

Historie
Hydroaminace je proveřená metoda výroby vůní z myrcenu. Při této reakci se diethylamin aduje na dienový substituent a katalyzátorem je diethylamid lithný.[2]
Roku 1989 popsal americký chemik Tobin Jay Marks vnitromolekulární hydroaminační reakce probíhající za přítomnosti odvozených od vnitřně přechodných kovů jako jsou lanthan, lutecium a samarium. Rychlosti reakcí se s rostoucími iontovými poloměry kovů snižovaly, což bylo patrně způsobeno sterickými jevy vytvářenými ligandy.[3] V roce 1992 Marks vyvinul první chirální katalyzátory hydroaminace, což umožnilo provádět stereoselektivní hydroaminační reakce.[4] První katalyzátor, který nebyl metalocenem, se objevil roku 2003; jeho součástí byly bisarylamidové a aminofenolové ligandy, což vedlo ke zvýšení enantioselektivity.[5]

Reaktanty a produkty
Hydroaminace byla vyzkoušena u mnoha různých aminů, primárních, sekundárních, cyklických, acyklických i anilinů, s rozdíknými sterickými a elektronovými vlivy; nenasycených substrátů (alkenů, alkynů a dienů) a také s několika rozdílnými katalyzátory. Také byly zkoumány vnitromolekulární hydroaminace několika různých aminoalkenů.[6]
Adice na nenasycenou vazbu uhlík-uhlík může, v závislosti na použitém katalyzátoru, proběhnout podle Markovnikovova pravidla i v rozporu s ním.[7] S ohledem na R/S chiralitu mohou vzniknout čtyři různé produkty: Markovnikovův produkt s R nebo S a neMarkovnikovův s R nebo S. I když byly popsány hydroaminace katalyzované mnoha různými kovy, tak je k selektivní syntéze jediného z těchto produktů možné použít jen menší část z nich. Byla popsána možnost připravit termodynamický nebo kinetický produkt.

Katalyzátory
Hydroaminaci může katalyzovat mnoho různých komplexů kovů jako jsou alkalické kovy, například lithium,[6] kovy 2. skupiny, jako je vápník,[8] či kovy 3. skupiny jako hliník,[9] indium[10] a bismut.[11] Byly také podrobně zkoumány možnosti využití katalyzátorů založených na přechodných kovech a lanthanoidech. Potenciální využití v této oblasti mají také zeolity.[6]
Mechanismus
Mechanismus hydroaminací katalyzovaných kovy je dobře znám.[12] Obzvlášť podrobně byla zkoumána vnitromolekulární hydroaminace alkenů katalyzovaná organickými sloučeninami lanthanoidů.[13]
Na začátku je katalyzátor aktivován výměnou amidové skupiny, čímž se vytváří aktivní katalyzátor (i). Následně se alken naváže na vazbu La-N (ii).[14] Nakonec dojde k protonolýze a tím ke vzniku cyklického produktu i regeneraci katalyzátoru (iii). I když byl tento mechanismus navržen pro katalyzátory obsahující lanthanoidy, tak velmi podobně funguje i u katalyzátorů založených na aktinoidech nebo alkalických kovech.
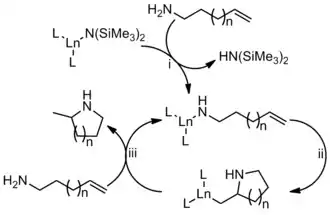
U katalyzátorů obsahujících přechodné kovy existuje několik modelů; první z nich spočívá v nukleofilním ataku alkenu nebo alkynu a jeho navázání na vazbu kov-amid.[12] Uvedené mechanismy jsou v souladu se studiemi reakční kinetiky, zkoumáním reakcí pomocí isotopového značkování i zachycováním předpokládaných meziproduktů.

Termodynamika a kinetika
Hydroaminační reakce jsou termodynamicky přibližně neutrální, mají však vysokou aktivační energii, což je pravděpodobně způsobeno vzájemným odpuzováním substrátu bohatého na elektrony a nukleofilního aminu. Vnitromolekulární reakce také mívají výrazně zápornou změnu entropie, a tak za vyšších teplot neprobíhají.[15][16] Z těchto důvodů jsou pro provádění těchto reakcí nutné katalyzátory.[17][12] Vnitromolekulární hydroaminace probíhají rychleji než mezimolekulární.
Termodynamické a kinetické produkty
Většina katalyzátorů hydroaminačních reakcí má dostatečnou účinnost pouze za vyšších teplot, obvykle tak vznikají pouze termodynamické produkty. Kinetické produkty byly izolovány a identifikovány například při tvorbě allylaminů z allenů. Směs allenů a derivátů anilinu byla za teploty 80 °C zahřívána za přítomnosti katalyzátoru obsahujícího rhodium.[18] V jiném případě byl použit katalyzátor obsahující palladium při pokojové teplotě s několika různými primárními i sekundárními, acyklickými i cyklickými aminy.[19] Oba tyto způspby provedení reakce mají vysokou výtěžnost; na násobnou vazbu lze následně připojovat další molekuly a vytvářet tak složitější sloučeniny.

Zásaditě katalyzované hydroaminace
Hydroaminace může být katalyzována silnými zásadami; příkladem je ethylace piperidinu pomocí ethenu:[20]
U některých takových reakcí se reaktivita s rostoucím počtem uhlíkových atomů alkenu anižuje.
Katalýza komplexy kovů 4. skupiny
Některé komplexy titanu a zirkonia mohou katalyzovat mezimolekulární hydroaminace alkenů a alkynů.[17] Poměrně podrobně byly prozkoumány reakce katalyzované zirconocen-bis(amido)komplexy. Titanocenamido- a titanocensulfonamidokomplexy katalyzují vnitromolekulární [2+2] cykloadiční hydroaminace aminoalkenů za vzniku příslušných azametallacyklobutanů. Jejich následnou protonolýzou se vytváří α-vinyl-pyrrolidinové (1) nebo tetrahydropyridinové (2) produkty. Výsledky teoretických a experimentálních studií jsou v souladu s navrhovanou tvorbou iminového meziproduktu.
Formální hydroaminace
Adice vodíku a aminové skupiny (NR2) za použití jiného činidla než aminu (HNR2) se označuje pojmem „formální hydroaminace“. I když tak nejsou k dispzici výhody například v podobě snadného zdroje dusíku, tak mohou být, díky lepším termodynamickým vlastnostem i možnostem použít velké množství aminačních činidel, také užitečné. Místo aminů se používají estery hydroxylaminu[21] a nitroareny.[22]
Využití
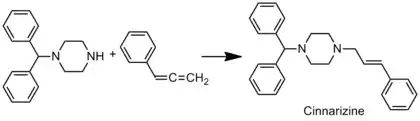
Hydroaminační reakce mají řadu využití díky využitelnosti aminů a šetrnosti k životnímu prostředí. Funkcionalizované allylaminy, které lze získat hydroaminací, mají mnoho možných použití ve farmaceutickém průmyslu. Hydroaminace lze použít k syntéze allylaminu cinnarizinu s vysokou výtěžností. Cinnarizinem je možné léčit závratě i kinetózu.[23]
Hydroaminaci je též možné použít k přípravě alkaloidů., příkladem je totální syntéza (-)-epimyrtinu.[24]
-epimyrtine.tif.png.webp)
Reference
V tomto článku byl použit překlad textu z článku Hydroamination na anglické Wikipedii.
- Hansjörg Grützmacher. Catalytic heterofunctionalization: from hydroanimation to hydrozirconation. Weinheim [u.a.]: Wiley-VCH, 2001. (1). ISBN 978-3527302345.
- Kunihiko Takabe; Takao Katagiri; Juntaro Tanaka; Tsutomu Fujita; Shoji Watanabe; Kyoichi Suga. Addition Of Dialkylamines To Myrcene: N,n-diethylgeranylamine. Organic Syntheses. 1989, s. 44.
- M. R. Gagné; Tobin Jay Marks. Organolanthanide-catalyzed hydroamination. Facile, regiospecific cyclization of unprotected amino olefins. Journal of the American Chemical Society. 1989, s. 4108.
- M. R. Gagné; L. Brard; V. P. Conticello; M. A. Giardello; T. J. Marks; C. L. Stern. Stereoselection effects in the catalytic hydroamination/cyclization of amino olefins at chiral organolanthanide centers. Organometallics. 1992, s. 2003.
- P. N. O'Shaughnessy; P. Scott. Biaryl amine ligands for lanthanide catalysed enantioselective hydroamination/cyclisation of aminoalkenes. Tetrahedron: Asymmetry. 2003, s. 1979.
- Thomas E. Müller; Matthias Beller. Metal-Initiated Amination of Alkenes and Alkynes. Chemical Reviews. 1998-04-01, s. 675–704. PMID 11848912.
- Matthias Beller; J. Seayad; A. Tillack; H. Jiao. Catalytic Markovnikov and anti-Markovnikov Functionalization of Alkenes and Alkynes: Recent Developments and Trends. Angewandte Chemie International Edition. 2004, s. 3368–3398. PMID 15221826.
- Mark R. Crimmin; Ian J. Casely; Michael S. Hill. Calcium-Mediated Intramolecular Hydroamination Catalysis. Journal of the American Chemical Society. 2005-02-01, s. 2042–2043. PMID 15713071.
- Jürgen Koller; Robert G. Bergman. Highly Efficient Aluminum-Catalyzed Hydro-amination/-hydrazination of Carbodiimides. Organometallics. 2010-22-11, s. 5946–5952.
- Rupam Sarma; Dipak Prajapati. Indium catalyzed tandem hydroamination/hydroalkylation of terminal alkynes. Chemical Communications. 2011-01-01, s. 9525–9527. Dostupné online. PMID 21776504.
- Kimihiro Komeyama; Yuusuke Kouya; Yuuki Ohama; Ken Takaki. Tandem ene-reaction/hydroamination of amino-olefin and -allene compounds catalyzed by Bi(OTf)3. Chemical Communications. 2011-01-01, s. 5031–5033. PMID 21423974.
- T. E. Müller; M. Beller. Metal-Initiated Amination of Alkenes and Alkynes. Chemical Reviews. 1998, s. 675–704. PMID 11848912.
- Sukwon Hong; Tobin J. Marks. Organolanthanide-Catalyzed Hydroamination. Accounts of Chemical Research. 2004-09-01, s. 673–686. PMID 15379583.
- Robert H. Crabtree. The organometallic chemistry of the transition metals. Hoboken, New Jersey: John Wiley, 2005. (4). Dostupné online. ISBN 978-0-471-66256-3.
- Jean-Jacques Brunet; Denis Neibecker; Francine Niedercorn. Functionalisation of alkenes: catalytic amination of monoolefins. Journal of Molecular Catalysis. 1989-02-01, s. 235–259.
- Adam M. Johns; Norio Sakai; André Ridder; John F. Hartwig. Direct Measurement of the Thermodynamics of Vinylarene Hydroamination. Journal of the American Chemical Society. 2006-07-01, s. 9306–9307. PMID 16848446.
- A. L. Reznichenko; K. C. Hultszch. Hydroamination of Alkenes. Organic Reactions. 2001, s. 1.
- Michael L. Cooke; Kun Xu; Bernhard Breit. Enantioselective Rhodium-Catalyzed Synthesis of Branched Allylic Amines by Intermolecular Hydroamination of Terminal Allenes. Angewandte Chemie International Edition. 2012-10-22, s. 10876–10879. PMID 23011801.
- John F. Beck; Danielle C. Samblanet; Joseph A. R. Schmidt. Palladium catalyzed intermolecular hydroamination of 1-substituted allenes: an atom-economical method for the synthesis of N-allylamines. RSC Advances. 2013-01-01, s. 20708–20718.
- J. WOLLENSAK AND R. D. CLOSSON. N-Methylpiperidine. Org. Synth.. 1973, s. 45. Dostupné online. (anglicky); Coll. Vol.. S. 575. (anglicky)
- Yuya Miki; Koji Hirano; Tetsuya Satoh; Masahiro Miura. Copper-Catalyzed Intermolecular Regioselective Hydroamination of Styrenes with Polymethylhydrosiloxane and Hydroxylamines. Angewandte Chemie International Edition. 2013-10-04, s. 10830–10834. ISSN 1521-3773. PMID 24038866.
- Jinghan Gui; Chung-Mao Pan; Ying Jin; Tian Qin; Julian C. Lo; Bryan J. Lee; Steven H. Spergel. Practical olefin hydroamination with nitroarenes. Science. 2015-05-22, s. 886–891. ISSN 0036-8075. PMID 25999503. Bibcode 2015Sci...348..886G.
- John F. Beck; Danielle C. Samblanet; Joseph A. R. Schmidt. Palladium catalyzed intermolecular hydroamination of 1-substituted allenes: an atom-economical method for the synthesis of N-allylamines. RSC Advances. 2013-01-01, s. 20708.
- Beilstein J. Org. Chem. 2013, 9, 2042–2047