Vrozená metabolická porucha
Vrozené metabolické poruchy (správněji dědičné metabolické poruchy (DMP)) představují heterogenní skupinu přibližně 800–900 genetických onemocnění, jejichž společným rysem je přítomnost biochemických nebo enzymatických odchylek, které je možné zjistit pouze speciálním vyšetřením. DMP jsou typickými představiteli skupiny vzácných nemocí („rare diseases“). Jsou děděné většinou autozomálně recesivním či gonosomálně recesivním i dominantním způsobem, u některých je dědičnost mitochondriální.[2] [3] Většina dědičných metabolických poruch je způsobena poškozením genu, který kóduje syntézu enzymu usnadňujícího přeměnu různých látek (substrátů) na látky jiné (produkty). Zdravotní potíže pacientů s těmito onemocněními vznikají buď v důsledku hromadění látek, které jsou pro organismus toxické či narušují jeho normální funkce, nebo kvůli snížené schopnosti syntetizovat pro organismus nezbytné sloučeniny. [4]
| Vrozená metabolická porucha | |
|---|---|
 Chlapec postižený Niemann-Pickovou chorobou | |
| Klasifikace | |
| MKN-10 | E70-E90 |
| MeSH | D008661 |
| Statistické údaje – obě pohlaví[1][2] | |
| Prevalence | 1 : 784 živě narozených dětí |
| Incidence | 1 : 500; 1 : 1500–5000 živě narozených dětí |
| Klinický obraz | |
| Průběh | chronický, akutní, nebo chronický s akutními atakami |
| Postižený systém | multiorgánové projevy v důsledku postižení různých metabolických drah |
| Některá data mohou pocházet z datové položky. | |
Historie
Počátky objevování dědičných metabolických poruch jsou spojeny se jménem Archibalda Garroda, který jako první poukázal na souvislost lidských nemocí a Mendelových zákonů dědičnosti a formuloval koncept dědičných metabolických poruch (inborn errors of metabolism). Garrod se zabýval studiem alkaptonurie a v roce 1902 publikoval knihu The Incidence of Alkaptonuria: a Study in Chemical Individuality, která je prvním záznamem případu recesivní dědičnosti u lidí. V roce 1923 vyšla další jeho kniha s názvem Inborn Errors of Metabolism, v níž publikoval své studie o alkaptonurii, cystinurii, pentosurii a albinismu. Právě překladem termínu inborn errors of metabolism vznikl u nás dlouho používaný nepřesný název vrozené metabolické poruchy (vrozené metabolické vady).
Etiopatogeneze
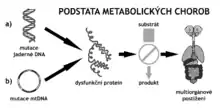
Příčinou DMP je geneticky podmíněná porucha funkce enzymu či transportního proteinu. Molekulovou podstatou této dysfunkce je obvykle homozygocie či smíšená heterozygocie u nemocí s autozomálně recesivním způsobem přenosu, hemizygocie u nemocí s gonozomálně recesivním přenosem a přítomnost mutací v nadkritickém množství organel u nemocí s mitochondriální dědičností. U části DMP jsou mutace v příslušných genech jediným postačujícím faktorem pro rozvoj klinicky patrné nemoci (především u nemocí organel). Naproti tomu u ostatních DMP je pro klinickou manifestaci (kromě genové mutace) nezbytné vystavení pacienta látce, kterou nedovede zpracovávat.
Důsledkem enzymové nebo transportní poruchy je hromadění substrátů příslušné reakce a chybění jejích produktů. Z nadbytečných substrátů navíc mohou vznikat toxické vedlejší produkty. Tyto komplexní změny v koncentraci metabolitů mohou mít nečekané a velmi vzdálené dopady na činnost buněk (např. na signální kaskády) a rovněž na zcela jiné orgány, než na ty, které jsou enzymovým deficitem postižené (např. u poruch cyklu močoviny jsou obvykle sídlem primárního defektu játra, nemoci se však projevují poruchami vědomí při systémové hyperamonemii).
Unikátní kombinace těchto dvou základních patogenetických mechanismů postihujících metabolismus látek s různou chemickou strukturou v rozmanitých orgánech pak vede ke vzniku celé škály klinických projevů lišících se u jednotlivých DMP. Změněné koncentrace metabolitů jsou obvykle prokazatelné ve tkáních a tělesných tekutinách, a proto je stanovení těchto metabolitů obvyklým diagnostickým postupem u DMP.[5]
Epidemiologie
V současné době již nelze vycházet z představy, že výskyt dědičných metabolických onemocnění v populaci je vzácný a že se s nimi praktický lékař během své praxe nesetká. Diagnóza metabolických onemocnění vyžaduje úzkou spolupráci mezi praktickými lékaři a specializovanými laboratořemi.[2][6]
Klasifikace
Pro seskupování jednotlivých DMP do vyšších hierarchických celků neexistují jednotná kriteria, různá klasifikační schémata vycházejí z různých aspektů DMP:[7]
- na základě subcelulární lokalizace nefunkčního proteinu (mitochondriální, lysozomální a peroxizomální poruhcy, poruchy Golgiho aparátu);
- definované metabolickou dráhou nebo typem metabolitu (např. poruchy metabolismu sacharidů, glykogenu, aminokyselin, lipidů);
- na základě společné analytické metodiky používané pro jejich průkaz (např. organické acidurie).[7]
- Z hlediska metabolitů, které způsobují klinické projevy onemocnění, lze dědičné metabolické poruchy rozdělit na nemoci malých molekul a nemoci velkých (komplexních) molekul.[2]
V následujícím textu bude použita klasifikace definovaná metabolickou dráhou nebo typem metabolitu.
- Poruchy metabolismu jednoduchých sacharidů - různé glycogen storage diseases
- Poruchy metabolismu aminokyselin - fenylketonurie atp.
- Poruchy metabolismu mastných kyselin mastných kyselin
- Poruchy intermediárního metabolismu
- Př. Klasické organické acidurie organic acid metabolism (organic acidurias) - např. alcaptonuria
- Mitochondriální nemoci, např. Kearnsův-Sayrův syndrom
- Poruchy metabolismu purinů a pyrimidinů - Lesch-Nyhanův syndrom
- Poruchy metabolismu porfyrinů
- Poruchy metabolismu komplexních sacharidů polysacharidů (glykoproteiny, proteoglykany, mukopolysacharidy, oligosacharidy)
- Poruchy metabolismu komplexních lipidů (sfingolipidy, mukolipidy)
- Poruchy metabolismu lipoproteinů
- Poruchy metabolismu steroidů
- Poruchy metabolismu peroxizomů
Klinické příznaky
Klinické příznaky metabolických onemocnění jsou různorodé a mohou se objevit u dětí i v pozdějším věku.[2][6][8] Jejich závažnost závisí na typu molekulárního postižení, stupni enzymového deficitu a funkci postiženého enzymu nebo jiného proteinu v metabolických dějích. V novorozeneckém a časném kojeneckém věku se manifestují závažné deficity klíčových enzymů a průběh těchto onemocnění je často smrtelný.[6]
Některá onemocnění se mohou projevit příznaky specifickými (typickými pro dané onemocnění), jindy se metabolická porucha projeví nespecificky (příznaky, které nacházíme i u jiných onemocnění).
- Mezi specifické příznaky patří například ektopie čočky a trombembolické příhody u homocystinurie.
- K nespecifickým příznakům se řadí poruchy psychomotorického vývoje, hypotonie, nechutenství, neprospívání. Teprve postupně dochází k rozvoji postižení funkce jednotlivých tkání (hypertrofická kardiomyopatie, demyelinizační postižení CNS, hepatomegalie, hepatopatie, katarakta, renální insuficience, atd.).
- Kombinací nespecifických příznaků někdy vzniká typický klinický obraz. Například u novorozence s výraznou hypotonií (snížení svalového napětí), klenutým čelem, širokým kořenem nosu, prostornou velkou i malou fontanelou, zvětšením jater a sleziny, poškozením jater a polycystickými ledvinami lze usuzoval na peroxizomální onemocnění.
Podezření na přítomnost metabolické poruchy by měly vzbudit zejména následující příznaky:
- nevysvětlitelná psychomotorická retardace, poruchy svalového napětí, křeče
- neobvyklý zápach potu a moče
- opakované epizody nejasného zvracení, acidózy, změny chování, poruchy vědomí
- hepatomegalie
- ledvinné kameny
- anamnestický údaj o konsanguinitě, opakovaných spontánních potratech, nevyjasněných úmrtích pod obrazem „sepse“, výskytu SIDS nebo atak Reye-like syndromu v rodině, by měly vždy vést k podezření na dědičné metabolické onemocnění.
Podle rychlosti nástupu klinických příznaků rozlišujeme metabolická onemocnění s akutním, intermitentním a chronickým průběhem.
- Akutní metabolická onemocnění se obvykle začínají projevovat již v novorozeneckém nebo raném kojeneckém věku, i když existují i pozdní formy těchto onemocnění. Často lze vysledovat provokující spouštěcí mechanismus rozvoje klinických příznaků. U poruch metabolismu aminokyselin, poruch v cyklu močoviny a organických acidurií dochází k projevům metabolického onemocnění po zahájení perorální výživy, při přechodu na stravu s vyšším obsahem bílkovin nebo v průběhu katabolismu při infektu. U poruch beta-oxidace mastných kyselin je to hladovění nebo nedostatečné krytí zvýšených energetických nároků organismu při zátěži (interkurentní infekce, stres, fyzická námaha, očkování). U každého donošeného fyziologického novorozence nebo malého kojence, u kterého dojde po bezpříznakovém období k prudkému zhoršení klinického stavu, kdy dítě přestává pít, zvrací, objevuje se porucha vědomí, křeče nebo respirační selhání, by mělo být vždy v rámci diferenciálně diagnostických úvah pomýšleno na dědičné metabolické onemocnění. Řada těchto onemocnění je léčebně ovlivnitelná včasným zahájením terapeutických postupů, mezi které patří například eliminace toxického metabolitu z organismu pomocí hemodiafiltrace, dietní opatření (nízkobílkovinná dieta, vyloučení některých složek ze stravy) nebo režimová opatření (antihypoglykemický režim).
- Metabolická onemocnění s intermitentním průběhem se manifestují v atakách, provokovaných změnou výživy nebo zvýšenou energetickou potřebou organismu v průběhu akutních stavů vyvolávajících katabolismus. Mezi atakami bývají nemocní bez jakýchkoliv klinických obtíží a také stanovení diagnosy je vázáno na období dekompenzace onemocnění (například přechodná forma leucinózy, opakované ataky Reye-like syndromu u některých poruch beta-oxidace mastných kyselin, pozdní formy deficitu ornitintranskarbamylázy – porucha v cyklu močoviny, deficit fruktózo-1,6-bisfosfatázy z poruch glukoneogeneze).
- Nejčastějším projevem chronicky progredujícího metabolického onemocnění je postupné zpomalení, zástava či dokonce regres původně normálního psychomotorického vývoje, nastupující po různě dlouhém bezpříznakovém období. Takto se začínají manifestovat střádavá lysozomální onemocnění ze skupiny mukopolysacharidóz a glykoproteinóz, u kterých se vyvíjí faciální dysmorfie s hrubými rysy v obličeji a projevy orgánového střádání (hepatosplenomegalie, zákaly rohovek, dysostosis multiplex, chlopenní vady, rozvoj hypertrofické kardiomyopatie). Neurodegenerativní metabolická onemocnění mají v klinickém obraze kromě deterioarace psychomotorického vývoje progredující neurologickou symptomatologii (Krabbeho choroba, metachromatická leukodystrofie, X-vázaná adrenoleukodystrofie, gangliosidózy, neuronální ceroidlipofuscinózy, Lesh-Nyhanův syndrom). Poruchy mitochondriálního energetického metabolismu se projevují zejména postižením funkce energeticky náročných tkání (CNS, srdce, svaly, játra) a všeobecnými příznaky (neprospívání, porucha růstu).[6][8]
Diagnostika
V případě dědičné metabolické poruchy způsobené deficitem enzymu nebo transportního proteinu dochází v místě metabolického bloku k hromadění specifických metabolitů. Podezření na určitou DMP lze pak vyslovit buď na základě stanovení zvýšené koncentrace metabolitů hromadících se nad metabolickýcm blokem nebo stanovením snížené koncentrace (event. nepřítomnosti) metabolitů pod tímto blokem. Podezření na DMP pak musí být ověřeno, což je možné provést na úrovni enzymu nebo genu.
Léčba
Moderní léčba pacientů s DMP je založená na znalosti patogeneze nemocí a závisí na typu choroby a její klinické závažnosti. V současné době je známa léčba přibližně u jedné třetiny pacientů s DMP.[2] V akutní fázi onemocnění se používá hemodialýza nebo hemodiafiltrace k odstranění toxických metabolitů z organismu.[2] Pro udržení dlouhodobé metabolické kompenzace u poruch metabolismu aminokyselin se používá nízkobílkovinná dieta doplněná o esenciální (nepostradatelné) aminokyseliny prostřednictvím speciálního dietního přípravku.[2] U poruch beta-oxidace mastných kyselin se doporučuje častá strava s omezeným obsahem tuků. V léčbě některých DMP se uplatňuje podávání vysokých dávek vybraných vitamínů.[2] U části pacientů s lysozomálním onemocněním je účinné injekční podávání rekombinantního enzymu, u jiných pacientů s tímto typem onemocnění se uplatňuje transplantace hematopoetickými kmenovými buňkami nebo orgánová transplantace (jater, ledvin, srdce).[9]
Prevence
Primární prevence' DMP není s ohledem na jejich genetický původ možná. Velký význam však má prevence sekundární, která spočívá ve včasné diagnostice konkrétní DMP. Klasickým příkladem úspěšné sekundární prevence onemocnění se včasně zahájenou příslušnou léčbou (pokud je léčitelné) jsou novorozenecké screeningové programy. S výjimkou nemocí s mitochondriální dědičností je naprostá většina DMP diagnostikovatelná v rizikových rodinách prenatálním vyšetřením.[2]
Odkazy
Reference
V tomto článku byly použity překlady textů z článků Inborn error of metabolism na anglické Wikipedii a Archibald Garrod na anglické Wikipedii.
- RAMACHANDRAN, Tarakad S. Inherited Metabolic Disorders Overview [online]. Rev. 2009-05-06 [cit. 2011-02-25]. Dostupné online. (anglicky)
- KOŽICH, Viktor; ZEMAN, Jiří. Dědičné metabolické poruchy v pediatrii. Postgraduální medicína. 9. 2010, roč. 12, čís. 7, s. 793–800. Dostupné online. ISSN 1214-7664.
- FERNANDES, J., et al. Diagnostika a léčba dědičných metabolických poruch. Praha: Triton, 2008. 607 s.
- Kolektiv autorů. Encyklopedie laboratorní medicíny pro klinickou praxi - verze 9 [databáze online]. Příprava vydání Antonín Jabor, Miroslav Zámečník, Luděk Straka. 9., aktualizované a doplněné vyd. Pardubice: SEKK, 2002-12-4, rev. 2009-12 [cit. 2011-03-25]. [Dále jen Encyklopedie laboratorní medicíny]. Dostupné v archivu pořízeném dne 2016-03-15. ISBN 8023897756.
- Encyklopedie laboratorní medicíny, Úvod do problematiky dědičných poruch metabolismu (Kožich, V.)
- Klinický obraz dědičných metabolických poruch (Zeman, J., Hrubá, E.)
- Úvod do problematiky dědičných poruch metabolismu (Kožich, V.)
- The Metabolic and Molecular Bases of Inherited Disease. Příprava vydání Charles R. Scriver, Artur L. Beaudet, William S. Sly. 8. vyd. USA: Mc Graw-Hill Education, 2002. 6000 s. ISBN 0-07-116336-0. (anglicky)
- BECK, M. Therapy for lysosomal storage disorders. IUBMB Life. 1. 2010, roč. 62, čís. 1, s. 33–40. Dostupné online. ISSN 1521-6551. (anglicky)
Literatura
- HOFFMANN, F.G.; ZSCHOCKE, J.; KAHLER MAYATEPEK, J.G.; MAYATEPEK, E. Dědičné metabolické poruchy. Praha: Grada, 2006. ISBN 80-247-0831-0. S. 416. (česky)
Externí odkazy
 Obrázky, zvuky či videa k tématu vrozená metabolická porucha na Wikimedia Commons
Obrázky, zvuky či videa k tématu vrozená metabolická porucha na Wikimedia Commons - (česky) Ústav dědičných metabolických poruch VFN a 1. LF UK (Praha)
- (česky) Laboratoř dědičných metabolických poruch FN a UP (Olomouc)
- (anglicky) Society for the Study of Inborn Errors of Metabolism (SSIEM)
- Výstižný název odkazu[nedostupný zdroj] – případné další informace
- Dokument pdf[nedostupný zdroj] – případné další informace
- https://web.archive.org/web/20030730063941/http://www.nupedia.com/%E2%80%93 internetová encyklopedie Nupedia (anglicky)
- – na Internet archive (anglicky)
- (anglicky) Journal of Inherited Metabolic Diseases
Přečtěte si prosím pokyny pro využití článků o zdravotnictví.