Fenylketonurie
Fenylketonurie (PKU) někdy nazývaná jako Føllingova nemoc je dědičné metabolické onemocnění spočívající v poruše přeměny aminokyseliny fenylalaninu na tyrosin, jenž u zdravých lidí katalyzuje jaterní enzym fenylalaninhydroxyláza (PAH). Právě mutaci genu kódujícího tento enzym má největší procento pacientů s fenylketonurií. V České republice populační frekvence výskytu fenylketonurie odpovídá 1: 9 000.
| Fenylketonurie | |
|---|---|
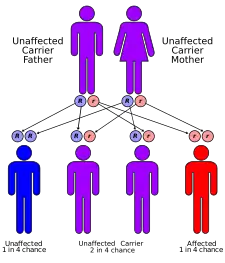 Fenylketonurie je dědičná autozomálně recesivně | |
| Klasifikace | |
| MKN-10 | E70.0 |
| MeSH | D010661 |
| Některá data mohou pocházet z datové položky. | |
Historie
První případy fenylketonurie byly zaznamenány již v roce 1934 u dvou sourozenců s těžkou mentální retardací. Důvod jejich postižení, poruchu metabolismu fenylalaninu, popsal norský lékař a biochemik Ivar Asbjorn Følling na základě izolace fenylpyruvátu z jejich moči. Pravou příčinu této choroby, mutaci jaterního enzymu fenylalaninhydroxylázy, následně určil mezi lety 1947 a 1953 lékař George Jervis. Od té doby bylo objeveno více než 280 dalších mutací tohoto i dalších enzymů hrající roli ve vzniku fenylketonurie.
Příznaky neléčené fenylketonurie
- Dítě postiženo touto chorobou začne brzy zaostávat, rozvíjí se mentální retardace
- Během 6. až 12. měsíce dochází k prvním epileptickým záchvatům, později až typu grand-mal, přičemž postižený nereaguje na léčbu antiepileptiky
- Dítě je neklidné, agresivní, apatické
- Na kůži se mohou vyskytovat ekzémy a vyrážky
- Postižený má bledou pleť, světlé vlasy a modré oči, což odráží nedostatek pigmentu melaninu, respektive jeho prekurzoru tyrosinu
Klasická a maternální fenylketonurie
Klasická fenylketonurie je autozomálně recesivní onemocnění – mutace genu kódující fenylalaninhydroxylázu se tak dědí od obou rodičů. Rozvoj příznaků postižení závisí především na včasnosti diagnostiky a nastolení speciální diety bez obsahu fenylalaninu. Z tohoto důvodu byl v roce 1975 v České republice zaveden povinný novorozenecký screening. Striktní dodržování diety je nezbytné v dětství, během něhož mají vysoké hladiny fenylalaninu toxický účinek na vyvíjející se mozek.
Maternální fenylketonurie je stav, kdy původně nepostižený potomek přebírá klinický obraz neléčené fenylketonurie matky (jedná o tzv. fenokopii). V důsledku vysokých koncentrací fenylalaninu v krevním oběhu pronikající skrze placentu dochází k nenávratnému poškození vyvíjejícího se plodu. Z tohoto důvodu je nutné, aby ženy s fenylketonurií plánující těhotenství opětovně zahájily dietu s nízkým obsahem fenylalaninu ještě před početím.
Variantní fenylketonurie a non-fenylketonurická hyperfenylalanemie
Obě tyto varianty fenylketonurie jsou zapříčiněny mutací fenylalaninhydroxylázy. Tedy stejného enzymu jako u klasické fenylketonurie, nicméně ten má v těchto případech částečnou aktivitu. V závislosti na velikosti zbytkové aktivity se odvíjí i léčba, která zahrnuje buď méně přísnou, nebo žádnou dietu. Dědičnost je opět autozomálně recesivní.
Defekty v metabolismu tetrahydrobiopterinu
Tetrahydrobiopterin je kofaktorem fenylalaninhydroxylázy. Během reakce přeměny fenylalaninu na tyrosin se jeho redukovaná forma 5,6,7,8-tetrahydrobiopterin oxiduje na 7,8-dihydropterin, který musí být opět redukován na aktivní kofaktor. Pokud dojde k nedostatečné syntéze či poruše opětovné redukce této látky, fenylalaninhydroxyláza není funkční a vyvíjí se fenylketonurie. Ta může být navíc v kombinaci s vrozenými neurologickými poruchami vyplývající z narušení metabolických drah katecholaminů či serotoninu, při jejichž syntéze se tetrahydrobiopterin rovněž účastní jako kofaktor. Odlišení poruchy v metabolismu tetrahydrobipterinu od klasické fenylketonurie má v dnešní době velký význam, vzhledem k tomu, že od roku 2009 je na trhu substituční lék, jež významně snižuje nesnášenlivost potravy s obsahem fenylalaninu.
Léčba
Léčba fenylketonurie spočívá především v dodržování striktní diety chudé na fenylalanin, a to především v dětském věku, kdy hrozí nenávratné poškození mozku a jeho funkce. Zároveň u těchto pacientů může daná dieta navodit deficienci některých esenciálních aminokyselin, minerálů či vitamínů, které je tak dobré nahrazovat vhodnými doplňky stravy.
Literatura
- HELLEKSON, Karen L. NIH Consensus statement on phenylketonuria. Am Fam Physician. 2001, 63, s. 1430–1432. Dostupný z www: http://www.aafp.org/afp/2001/0401/p1430.html
- NUSSBAUM, Robert L.; MCINNES, Roderick R.; WILLARD, Huntington F. Thompson and Thompson: Klinická genetika. 1. vydání. Praha: Triton, 2004. 426 s. ISBN 80-7254-475-6.
- ŠŤASTNÁ, Sylvie et al. Přehled vyšetření metabolitů pro diagnostiku dědičných metabolických poruch. 1. vydání. Praha: Ústav dědičných metabolických poruch VFN a 1. LF UK Praha, 2008. 92 stran. ISBN 978-80-904219-0-5
- VOET, Donald; VOETOVÁ, Judith G. Biochemie. 1. vydání. Praha: Victoria Publishing, 1995. 1325 s. ISBN 80-85605-44-9.
- WERNER, Ernest R. et al. Tetrahydrobiopterin: biochemistry and pathophysiology. Biochem. J. 2011, 438, s. 397–414 Dostupný z www: http://www.biochemj.org/bj/438/0397/bj4380397.htm
- WIDAMAN, Keith F. Phenylketonuria in Children and Mothers: Genes, Environments, Behavior. Curr Dir Psychol Sci. 2009, 18, s. 48–52. Dostupný z www: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2705125/?tool=pubmed
- WILLIAMS, Robin A. et al. Phenylketonuria: An Inborn Error of Phenylalanine Metabolism. Clin Biochem Rev. 2008, 29, s. 31–41. Dostupný z www: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2423317/?tool=pubmed
Externí odkazy
 Obrázky, zvuky či videa k tématu fenylketonurie na Wikimedia Commons
Obrázky, zvuky či videa k tématu fenylketonurie na Wikimedia Commons