Retinoblastom
Retinoblastom je druhým nejrozšířenějším zhoubným nádorem oka (hned po uvealním malignomu) a vůbec nejrozšířenější dětská oční malignita. Jedná se o autosomálně dominantní onemocnění, kdy u méně než 10 % pacientů nacházíme pozitivní rodinou anamnézu. Nejčastěji se vytváří z buněk zárodečné sítnice oka, které se v pozdějších fázích nitroděložního vývoje diferencují na fotoreceptory. Metastatickými ložisky může být poseta i celá sítnice.

Retinoblastom mívá bělavou barvu, způsobenou především kalcifikacemi v jeho nekrotické tkáni. Pod mikroskopem jsou patrné rozetovité, až kopretinovité formace buněk.
Vznik a druhy retinoblastomu
Retinoblastom je dáván do souvislosti s poruchou genu RB1 na dlouhém raménku 13. chromozomu, ať už získanou nebo zděděnou. V každé buňce sítnice jsou přítomny dvě alely pro gen RB1 (což je gen pro tzv. retinoblastomový protein). Dříve, nežli může vzniknout retinoblastom, musí být vyřazeny nebo mutovány obě alely tohoto supresorového genu (tj. genu, který potlačuje nekontrolovatelné dělení).
Retinoblastom se vyskytuje ve dvou formách, dědičné (predispoziční, výjimečné) a nedědičné. Dědičná forma se objevuje asi u 40 % pacientů s retinoblastomem, zbývajících 60 % má formu nedědičnou. Podle toho, zda zasáhne obě nebo jedno oko, ho pak také rozdělujeme na bilaterální nebo unilaterální.
- Při nedědičné formě musí postupně v jedné jediné buňce sítnice vzniknout dvě mutace (aby byly poškozeny obě alely pro gen RB); onemocnění je dále nepřenosné, protože mutace je pouze v tělní buňce a nikoliv v pohlavní linii. Děti s touto formou mají prakticky výlučně jediný retinoblastom (v jednom oku), který se objeví nejčastěji okolo druhého roku života.
- Dědičná forma postihuje zpravidla obě oči a objevuje se v dřívějším věku. Pokud však retinoblastom postihne pouze jedno oko, bývá klinicky obtížné přesně odlišit obě formy od sebe. Co se predispoziční formy týče, jedna ze dvou potřebných mutací je přítomna již v zárodečné buňce (tato mutovaná alela je buď zděděna od jednoho z rodičů, nebo vzniká při prenatálním vývoji) a bude tedy přítomna ve všech buňkách, které z ní vzniknou. Poté stačí, aby došlo již jen k jedné jediné mutace, a protože cílových buněk oční sítnice je kolem deseti milionů, retinoblastom je téměř vždy mnohočetný a postiženy bývají obě oči. Proto je i vznik časnější, někdy je retinoblastom přítomen již při narození.
Někdy se popisuje trilaminární forma spojená s postižením šišinky.
Diagnostika
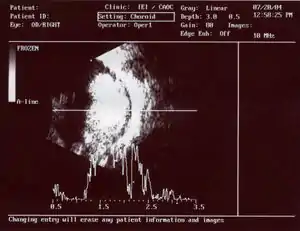
Prvními příznaky onemocnění bývá zpravidla šilhání (strabismus) nebo leukokorie, objevuje se i otok a zvětšení oka nebo zarudnutí v jeho okolí. U postižených dětí se také často objevuje zvláštní bělavý odraz v očích na fotografiích. Světlá skvrna v oku se dá lépe pozorovat za horších světelných podmínek, kdy dochází k rozšíření zornice oka. Dědičné formy retinoblastomu jsou často objeveny lékaři již po porodu. Průběh onemocnění se dělí do pěti stádií podle závažnosti, pokud je nádor brzo podchycen, zvyšuje se šance na vyléčení.
V ČR byl v roce 2005 zaveden screening novorozenců právě na leukokorii (nevybavení červeného reflexu při posvícení do oka oftalmoskopem). Vyšetření provádí zaměstnanci porodnice. Pokud se nepodaří červený reflex vyvolat, je pozván na konzilium oftalmolog. Do dif. dg. leukorie patří vrozená katarakta, perzistující primární sklivec či larvální toxokaróza. Při ultrazvukovém vyšetření je za nádorem sonografický stín díky kalcifikacím, které se zpravidla v tumoru objeví.
Průběh
Retinoblastom se šíří choroidální invazí s následnou hematogenní diseminací a cestou nervus opticus do subarachnoideálního prostoru (schopnost zasahovat do zrakového nervu a metastázovat až do mozku pacienta). Pokud se podaří pacienta vyléčit, tak u predispoziční formy často vznikají osteosarkomy. V rozvojových zemích je však toto onemocnění často smrtelné. Pokud se léčba nedaří, lékaři přistupují k odstranění postiženého oka.
Terapie
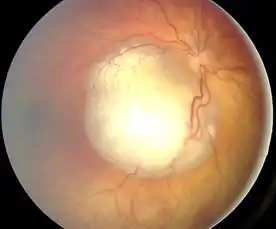
Retinoblastom se dá léčit mnoha druhy radioterapie, chemoterapie a dalšími postupy, které se pro urychlení a usnadnění léčby často kombinují:
- enukleace – nejstarší způsob léčby, kdy je odstraněna celá oční koule, dnes vyhrazený pouze pro některé případy – zvláště pokud nezabírá léčba cytostatiky
- teleterapie – je vysoce radiosenzitivní
- brachyterapie – ozařování radioaktivní plombou, malý radioaktivní implantát se umístí na bělimu přímo do oka pacienta nad bází nádoru a ozařuje nádorové ložisko, využívá se hlavně jako sekundární léčba retinoblastomu
- laserová fotokoagulace – koagulace cév zásobujících nádorové ložisko
- kryoterapie – využívá se k ošetření okrajových částí nádoru
- transpupilární termoterapie – zvýšením teploty na rozmezí 42° až 60° dojde k poškození nádorové tkáně, ale zachování retinálních cév
- intravenózní chemoredukce – chemoterapie využívající zpravidla vepesid a karboplatinu, zmenšuje nádor a umožňuje zachovat celé oko pacienta
- termochemoterapie – kombinace termoterapie a chemoterapie
- genová terapie – nová perspektivní metoda léčby, stále ve výzkumu
- lokálně destruktivní léčba – u malých a příznivě lokalizovaných nádorů
Odkazy
Literatura
- Bc. Punko, A. Dlouhodobé funkční výsledky komplexní léčby retinoblastomu. Brno: Masarykova univerzita, Lékařská fakulta, 2009. Dostupné online
- MUDr. Pochop, P. Nádory oka a očnice v dětském věku. VOX PEDIATRIE, 2003, č. 8, ročník 3.
- Kuchynka, P. et all. Oční lékařství: Souček, P. Nitrooční nádory. Grada, 2007. Dostupné online
Externí odkazy
 Obrázky, zvuky či videa k tématu retinoblastom na Wikimedia Commons
Obrázky, zvuky či videa k tématu retinoblastom na Wikimedia Commons - Český web o retinoblastomu u dětí
- Společnost Retinoblastoma international - anglicky Archivováno 3. 2. 2012 na Wayback Machine
- Retinoblastoma.com - anglicky
- Retinoblastom na OFTA - Oční laserové centrum a mikrochirurgie oka.
- Retinoblastoma brochure: Retinoblastoma international - anglicky. Archivováno 18. 10. 2011 na Wayback Machine
- Komplexní onkologické centrum.[nedostupný zdroj]
- Laboratoř molekulární diagnostiky. FN Brno.
- Současné terapeutické postupy u retinoblastomu. Zdravotnické noviny, 2000.[nedostupný zdroj]
Přečtěte si prosím pokyny pro využití článků o zdravotnictví.