Retinoblastomový protein
Retinoblastomový protein (pRb, RB1, RB1) je DNA-vazebný protein přítomný v jádře savčích buněk, který hraje významnou roli v regulaci transkripčních faktorů. Díky této roli je schopen tlumit buněčné dělení[1] a tedy i rakovinné bujení – patří mezi nejznámější tumor supresorové geny.[2][3]
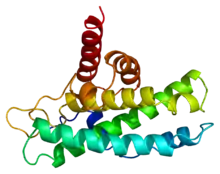
Stavba
Rb protein je složen z 928 aminokyselin[4] a má molekulovou hmotnost 110 kDa.[1] Skládá se ze tří domén, centrální doména se označuje jako kapsa a skládá se ze dvou subdomén připomínajících cyklinový „box“. Velmi podobný motiv je, opět ve dvou kopiích, v RBN oblasti na N-konci. C-koncová část nemá pevnou strukturu a je tedy jedinou rozsáhlejší částí proteinu Rb, která se nepodílí na jeho modulárním uspořádání.[4]
Funkce
Geny z rodiny Rb (do níž mimo Rb patří ještě geny p107 a p130[5]) hrají roli v širokém spektru buněčných dějů, ale nejlépe jsou známy jako regulátory buněčného cyklu savců. V G1 fázi se totiž váží na E2F jaderné transkripční faktory a s pomocí jistých chromatin modelujících proteinů zastavují transkripci mnohých cílových genů. Tento účinek zjednodušeně brání buňkám, aby se nekontrolovaně množily. Signál k buněčnému dělení dávají cyklin-dependentní kinázy (CDKs), které v pravou chvíli vyvazují Rb a ten se díky mnohočetné fosforylaci stává neaktivním. Dochází k transkripci např. cyklinů typu E a A, které směřují buňku z G1 do S fáze. K opětovné aktivaci Rb dochází (defosforylací určitými fosfatázami) až během následující mitózy.[5]
Poruchy
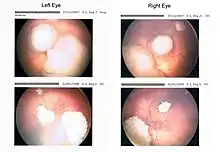
Mutantní gen RB je příčinou tzv. retinoblastomu, zhoubného nádoru oka. Může být dědičné i nedědičné povahy a postihuje osoby často již v dětském věku. Mutace RB v čípcích vede k uvolnění některých transkripčních faktorů, buňky snadno opouští G1 fázi buněčného cyklu a směřují do buněčného dělení. To v oku způsobuje nádorové bujení buněk sítnice a tedy vznik nádoru.[3]
Mutace v genu RB však mohou u některých jedinců (včetně osob s vyléčeným retinoblastomem oka) vést i k jiným nádorům. U jedinců s mutantním RB genem byly zaznamenány kostní nádory nebo rakovina močového měchýře, ale také rakovina prsu, plic či prostaty, stejně jako akutní myeloidní leukemie. Záleží zejména na tom, v jaké části těla k mutaci RB došlo.[3]
Reference
- Oxford dictionary of biochemistry and molecular biology; revised edition. Příprava vydání R. Cammack et al. New York: Oxford university press, 2006. ISBN 0-19-852917-1.
- MCCLEAN, Phillip. Tumor Suppressor Genes [online]. 1997 [cit. 2013-06-24]. Dostupné online.
- LEWIS, Ricki. Human Genetics: Concepts and Applications. 9. vyd. [s.l.]: McGraw−Hill, 2009. Dostupné online. S. 365.
- DICK, Frederick A., Rubin, Seth M. Molecular mechanisms underlying RB protein function. Nature Reviews Molecular Cell Biology. NaN-NaN-NaN, roč. 14, čís. 5, s. 297–306. DOI 10.1038/nrm3567.
- GORDON, Gabriel M., Du, Wei. Conserved RB functions in development and tumor suppression. Protein & Cell. 2011-12-17, roč. 2, čís. 11, s. 864–878. DOI 10.1007/s13238-011-1117-z.