IPEX
IPEX (z angl. immunodysregulation polyendocrinopathy enteropathy X-linked) syndrom je monogenně podmíněná choroba způsobená mutací v genu FOXP3. FOXP3 je známým transkripčním faktorem zásadním pro vývoj T regulačních lymfocytů. Právě dysfunkce T regulačních lymfocytů je příčinou IPEX syndromu. Nejběžnějšími příznaky jsou poruchy imunitní regulace ve formě autoimunitní enteropatie, dermatitidy a endokrinních žláz. Onemocnění se projevuje už v prvních měsících života a v případě zanedbání včasného odhalení a léčebné intervence může být fatální. Jedinou dosud známou léčbou je transplantace hematopoetických kmenových buněk.[1][2]
Symptomy
Jak už zkratka anglického názvu napovídá, IPEX syndrom se manifestuje nejběžněji v orgánech spjatých s imunitním a endokrinním systémem. Nejčastěji mívají pacienti problémy jako:
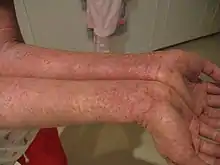
- dermatitida a ekzémy,
- průjmová onemocnění,
- hemolytická anémie, trombocytopenie nebo jiné hematologické abnormality,
- diabetes mellitus 1. typu,
- autoimunitní thyroiditida (porucha štítné žlázy).
Častým příznakem je také to, že dítě dobře neprospívá.
Po první roce života se nemoc může navíc projevovat autoimunitní hepatitidou nebo nefropatií.
Nejčastěji se první příznaky objevují v době od narození po prvních 7 měsíců věku. Existují ale i případy s pozdějším nástupem. [3][4]
IPEX syndrom byl poprvé popsán v roce 1982 na případu rodiny, v níž během prvních letech života zemřelo 11 chlapců na komplikace imunitní povahy po očkování. Dalších 8 chlapců v rodině mělo obdobné problémy s náchylností k infekcím a také s autoimunitními patologiemi. Chlapci trpěli průjmy, ekzémy, diabetem 1. typu a autoimunitní postižením štítné žlázy. Pouze dva z nich přežili první dekádu svého života.[5]
Genetické příčiny
Mutace v genu FOXP3 vedoucí k expresi nefunkčního proteinu se nejběžněji nachází v oblasti, která je zodpovědná za vazbu transkripčního faktoru na DNA. Konkrétně se jedná o oblast forkhead domény, podle které dostal tento gen a stejnojmenný protein i své jméno – Forkhead Box Protein 3. Právě fakt, že je u pacientů s IPEX syndromem často mutovaná DNA vazebná doména a FOXP3 nemůže plnit svou funkci transkripčního faktoru, je příčinou imunitní dysregulace při vývoji a funkci T regulačních lymfocytů. Jejich absence nebo dysfunkce způsobují typické symptomy autoimunitní povahy u IPEX syndromu.[1]
Momentálně (data dostupná k roku 2018) se uvádí kolem 70 popsaných nemoc způsobujících mutací u člověka, ale toto číslo se rychle mění.[6] Např. v roce 2010 bylo známo pouze okolo 20 mutací FOXP3 genu způsobujících IPEX syndrom.[1]
Gen FOXP3 se nachází na pohlavním chromozomu X v oblasti Xp11.23. Lokalizací na pohlavním chromozomu je daná i dědičnost onemocnění, která je X-vázaná gonozmálně recesivní.[7]
Výzkumný myší model
IPEX syndrom jako jedno z mála jasně monogenně podmíněných imunitních onemocnění je možné zkoumat i pomocí speciálního myšího modelu, který imituje průběh onemocnění u lidí. Jsou to tzv. scurfy myši, které mají v genu FOXP3 zárodečně vnesenou mutaci. Konkrétně se jedná o inzerci o velikosti 2 nukleotidových bazí, která vede k expresi nefunkčního proteinu. U scurfy myší můžeme typicky najít zvětšenou slezinu a mízní uzliny, červené oči a problémy kožní povahy. Tyto myši poté velmi brzy na následky onemocnění umírají – uvádí se cca po 3 týdnech života.[6]
Léčba
Jedinou možnou známou léčbou je transplantace hematopoetických kmenových buněk.
Jako podpůrná terapie se ale také standardně užívá podávání imunosupresivních léčiv. Konkrétně se jedná o steroidní látky, inhibitory kalcineurinu (takrolimus, cyklosporin A) a non-kalcineurinové inhibitory. (rapamycin, methotrexát, mykofenolát mofetil aj.). Také se využívá biologické léčby ve formě podávání monoklonálních protilátek proti TNF alfa nebo CD20.[4]
Odkazy
Reference
- MICHELS, Aaron W.; GOTTLIEB, Peter A. Autoimmune polyglandular syndromes. Nature Reviews Endocrinology. 2010-5, roč. 6, čís. 5, s. 270–277. Dostupné online [cit. 2019-06-29]. ISSN 1759-5029. DOI 10.1038/nrendo.2010.40. (anglicky)
- BARZAGHI, Federica; PASSERINI, Laura; BACCHETTA, Rosa. Immune Dysregulation, Polyendocrinopathy, Enteropathy, X-Linked Syndrome: A Paradigm of Immunodeficiency with Autoimmunity. Frontiers in Immunology. 2012, roč. 3. Dostupné online [cit. 2019-06-29]. ISSN 1664-3224. DOI 10.3389/fimmu.2012.00211.
- GISCHEL, Melissa E.; BECK, Carolyn; HALL, Margot. A Mystery Diagnosis: Immune Dysregulation, Polyendocrinopathy, Enteropathy, X-Linked Recessive. Laboratory Medicine. 2009-5, roč. 40, čís. 5, s. 303–306. Dostupné online [cit. 2019-06-30]. ISSN 0007-5027. DOI 10.1309/LMCXCU6GZE7G8ZEI. (anglicky)
- BARZAGHI, Federica; AMAYA HERNANDEZ, Laura Cristina; NEVEN, Benedicte. Long-term follow-up of IPEX syndrome patients after different therapeutic strategies: An international multicenter retrospective study. Journal of Allergy and Clinical Immunology. 2018-3, roč. 141, čís. 3, s. 1036–1049.e5. Dostupné online [cit. 2019-06-30]. DOI 10.1016/j.jaci.2017.10.041. (anglicky)
- POWELL, Berkley R.; BUIST, Neil R.M.; STENZEL, Peter. An X-linked syndrome of diarrhea, polyendocrinopathy, and fatal infection in infancy. The Journal of Pediatrics. 1982-5, roč. 100, čís. 5, s. 731–737. Dostupné online [cit. 2019-06-30]. DOI 10.1016/S0022-3476(82)80573-8. (anglicky)
- BACCHETTA, Rosa; BARZAGHI, Federica; RONCAROLO, Maria-Grazia. From IPEX syndrome to FOXP3 mutation: a lesson on immune dysregulation: IPEX syndrome and FOXP3. Annals of the New York Academy of Sciences. 2018-4, roč. 1417, čís. 1, s. 5–22. Dostupné online [cit. 2019-06-30]. DOI 10.1111/nyas.13011. (anglicky)
- OMIM Entry - * 300292 - FORKHEAD BOX P3; FOXP3. www.omim.org [online]. [cit. 2019-06-30]. Dostupné online.
Přečtěte si prosím pokyny pro využití článků o zdravotnictví.