Enová reakce
Enová reakce (také Alderova enová reakce, jelikož ji objevil Kurt Alder) je reakce alkenu s allylovým vodíkem (enem) a sloučeniny obsahující násobnou vazbu (enofilem) a vzniká tak další vazba σ přesunem dvojné vazby enu a přemístěním vodíku mezi pozicemi 1 a 5. Produktem je substituovaný alken s dvojnou vazbou posunutou do allylové pozice.[1]
Enové reakce jsou druhem pericyklických reakcí[2] a tak jsou k jejich provedení často potřeba vysoce aktivované substráty a nebo vysoké teploty.[3] Při reakci ovšem může ovšem být na enu i enofilu připojeno mnoho různých funkčních skupin Bylo vyvinuto též mnoho enových reakcí katalyzovaných Lewisovými kyselinami, díky čemuž lze dosáhnout vysoké výtěžnosti a selektivity při mnohem nižších teplotách, což z enové reakce činí užitečný postup tvorby vazeb C-C při přípravě složitějších molekul.
Enový reaktant
Eny jsou látky s molekulami alespoň jednu vazbu pí a jedním či více aktivními vodíkovými atomy na allylové, propargylové nebo v α pozici. Skupiny v enech mohou být například alkenové, alkynové, allenové, aromatické, cyklopropylové a také vazby uhlík-heteroatom.[4] Enové reakce se obvykle účastní allylový vodík allenových sloučenin, avšak u allenylsilanů dochází k přesunu allenového vodíku v poloze α vůči substituentu obsahujícímu křemík, čímž se vytváří silylalkyn. Enovou složkou reakce může být také fenol; příkladem může být reakce s dihydropyranem, která ovšem proníhá za vysokých teplot (150–170 °C). Eny s velkým úhlovým napětím a malými cykly reagují i za výrazně nižších teplot.
Byly též popsány eny se skupinami C=O, C=N a C=S, ovšem objevují se vzácně.[4]
Enofilový reaktant
Enofily jsou molekuly s násobnými vazbami, které mají substituenty snižující elektronovou hustotu. Může jít o sloučeniny s násobnými vazbami mezi atomy uhlíku, mezi atomem uhlíku a heteroatomem (například C=O, C=N, C=S, C≡P) mezi dvěma heteroatomy (N=N, O=O, Si=Si, N=O, S=O), kumulenovými systémy (N=S=O, N=S=N, C=C=O, C=C=S, SO2) a nebo elektricky nabitými systémy (C=N+, C=S+, C≡O+, C≡N+).[4]
Mechanismus
Soustředěný mechanismus a přechodné stavy
Hlavní interakcí hraničních orbitalů objevující se při enové reakci je ta mezi HOMO enu a LUMO enofilu.[5] HOMO enu vzniká kombinací orbitalu vazby pí ve vinylové skupině a orbitalu vazby mezi uhlíkem a allylovým vodíkem. Všechny uhlíkovo-enové reakce mají vysokou aktivační bariéru, při reakcích propenu a ethenu je to asi 138 kJ/mol.[6] S rostoucí polaritou (od ethanu k formaldehydu) enofilu má ovšem jeho LUMO větší amplitudu na uhlíku, což vede k většímu překryvu C-C a menšímu překryvu H–O, kvůli čemuž reakce probíhá asynchronně. Při nahrazení kyslíku sírou v molekule enofilu se aktivační bariéra snižuje na přibližně 61,5 kJ/mol. Zkoumáním aktivačních bariér a aktivačních napětí různých enových reakcí s propenem jako enovým reaktantem bylo zjištěno, že se aktivační bariéry postupně snižují v řadě H2C=CH2 > H2C=NH > H2C=CH(COOCH3) > H2C=O > H2C=PH > H2C=S a v této řadě se také reakce stává stále asynchronnější a aktivační napětí se postupně snižuje.[6]

Uvedené vlastnosti tohoto typu reakce byly ověřeny experimentálně[7] a reakce byla ve Woodwardově notaci označena jako [σ2s + π2s + π2s].[5] Jako meziprodukt tepelné enové reakce propenu s formaldehydem byla nejprve navržena molekula s obálkovou konformací a úhlem C–O–H o velikosti 155°.[8]
V roce 2011[9] byly prozkoumány nekatalyzhované vnitromolekulární karbonylo-enové reakce používané na přípravu cyklopentanových fragmentů přírodních i umělých jatrofa-5,12-dienů, což je skupina P-glykoproteinových modulátorů. Výpočetními metodami bylo zjištěno, že reakce může probíhat přes dva různé spojené meziprodukty s obálkovými či podobnými konformacemi. Selektivita tohoto procesu se vysvětluje vlivem 1,3-transanulárních interakcí.
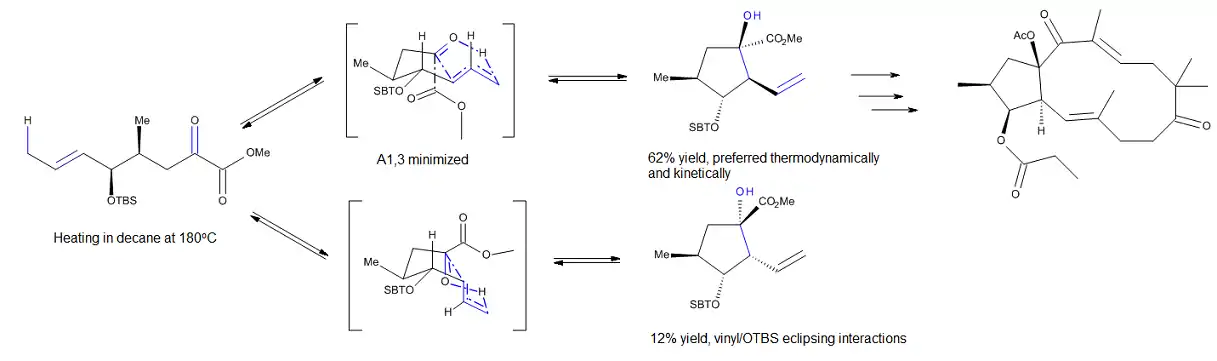
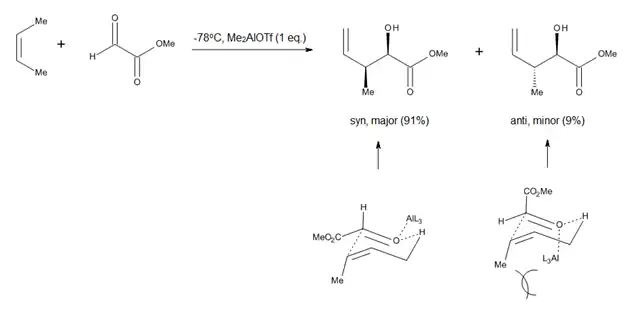
Studium karbonylo-enových reakcí probíhajících za přítomnosti Lewisových kyselin, jako například glyoxylát-enových reakcí katalyzovaných hliníkem, vedlo k domněnkám, že židličkové a podobné konformace zaujímají až později vznikající meziprodukty enových reakcí.[10]
Radikálový mechanismus
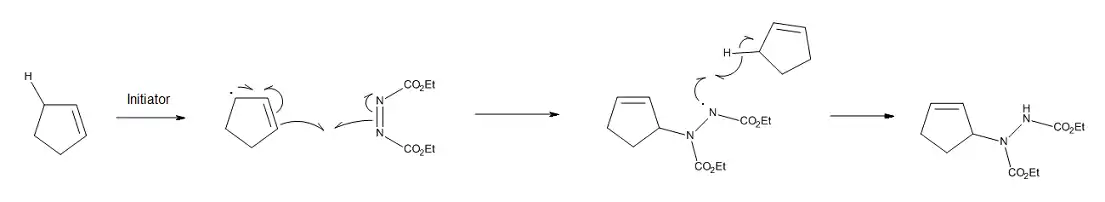
Pokud je reakce podle soustředěného mechanismu geometricky nevýhodná, tak může tepelná enová reakce probíhat biradikálově[11] nebo, pokud se v reakční směsi nacházejí iniciátory radikálů, přes volné radikály. Jako příklad lze uvést reakce cyklopentenu a cyklohexenu s diethylazodikarboxyláty, které mohou být katalyzovány iniciátory radikálů. Postupný průběh reakce je, jak je znázorněno na následujícím obrázku, podpořen cyklopentenylových a cyklohexenylových radikálů a také tím, že cyklopenten a cyklohexen by při soustředěném mechanismu obtížně zaujímaly ideální geometrii.[12]
Regioselektivita
Podobně jako u každé cykloadice úspěšnost enové reakce značně závisí na sterické dostupnosti allylového vodíku u enu. Obecně platí, že methylové a methylenové atomy vodíku se odstraňují snadněji než methynové vodíky. U tepelných enových reakcí reaktivita klesá v řadě primární skupina > sekundární skupina > terciární skupina, což odpovídá stabilitě alkenového meziproduktu. Při reakcích katalyzovaných Lewisovými kyselinami snadnost odtržení methylového či methylenového vodíku značně ovlivňuje dvojice enofil/Lewisova kyselina.[10]
Orientaci adice enu lze určit pomocí relativní stabilizace vznikajících částečných nábojů v nesymetrickém meziproduktu při tvorbě vazeb σ. Více zastoupený stereoizomer vznikne z přechodného stavu, u kterého jsou částečné náboje orientací enu a enofilu stabilizovány nejlépe.[4]
Vnitřní asymetrická indukce
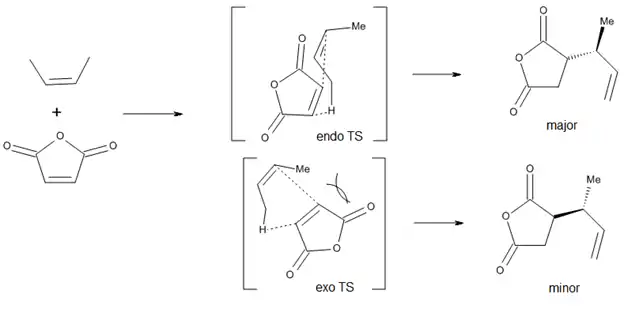
Při zkoumání diastereoselektivity s ohledem na nově vznikající chirální centra byly kvalitativně pozorovány endo produkty, tuto preferenci ovšem mohou pozměnit sterické efekty.[10]
Vnitromolekulární enové reakce
Vnitromolekulární enové reakce mají nižší zápornou entropii aktivace, díky čemuž se často snadněji provádějí, dochází k nim i u jednoduchých enofilů, jako jsou neaktivované alkeny a alkyny.[13] Vysoká regioselektivita a stereoselektivita těchto reakcí poskytuje možnost připravovat složité cyklické molekuly.
V závislosti na poloze skupiny spujující en a enofil Oppolzer[10] rozdělil tepelné a Lewisovými kyselinami katalyzované vnitromolekulární enové reakce do tří druhů (I, II a III) a Snider[3] přidal další druh (IV) (viz následující obrázek). Při těchto reakcích je překryv orbitalů enu a enofilu ovlivňován převážně geometrií přiblížení obou reaktantů.
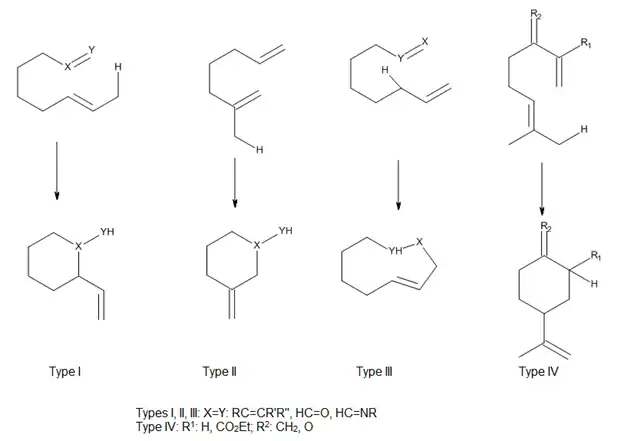
Enové reakce katalyzované Lewisovými kyselinami
Tepelné enové reakce mají řadu nevýhod jako jsou potřeba vysokých teplot a vedlejší reakce, například protony katalyzované polymerace alkenů nebo izomerační reakce. Jelikož mají enofily nedostatek elektronů, tak jejich spojení s Lewisovými kyselinami reakci urychluje:
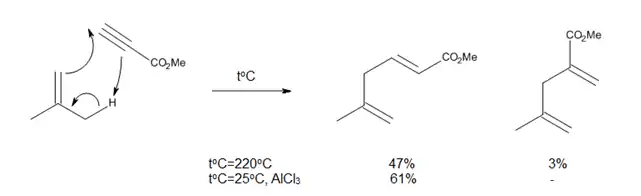
K zachytávání protonů se nejčastěji používají alkylaluminiové halogenidy, které rozšiřují možnosti enových reakcí a umožňují jejich zkoumání za mírnějších podmínek.[3]
Lewisovy kyseliny mohou přímo vytvořit komplex s karbonylovým kyslíkem a proto bylo vyvinuto mnoho trialkylaluminiových katalyzátorů obsahujících vazbu C=O. Bylo zjištěno, že (CH3)2AlCl je velmi vhodným katalyzátorem enových reakcí α,β alifatických, především nenasycených, aldehydů a ketonů; tuto vlastnost má díky tomu, že příslušný komplex dále reaguje za vzniku methanu a hlinitého alkoxidu, což může zabránit přesmykům katalyzovaným protony a solvolýze, jak je znázorněno na následujícím obrázku:[3]
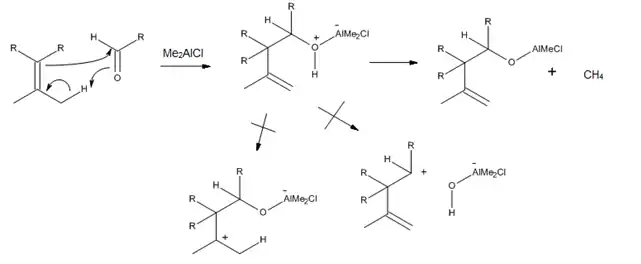
Při řízených karbonyl–enových reakcích byla pozorována vysoká regioselektivita a stereoselektivita po přidání Lewisovy kyseliny, což lze vysvětlit pomocí meziproduktů s židličkovitými či podobnými konformacemi. Některé z těchto reakcí mohou probíhat za velmi nízkých teplot a stále přitom poskytovat velmi dobré výnosy jednoho stereoizomeru.[10]
Reakční podmínky
Katalytická množství Lewisových kyselin jsou užitečná při mnoha enových reakcích s reaktiními enofily, protože omezují vedlejší reakce. Potřebné množství Lewisovy kyseliny se může značně lišit, jelikož záleží na realtivní zásaditosti enofilu a enového adduktu. Z hlediska rozpouštědel bývají největší výtěžky dosaženy při použití halogenovaných uhlovodíků jako rozpouštědel; polární rozpouštědla jako ethery nejsou vhodná, protože tvoří s Lewisovými kyselinami komplexy a tím je jakožto katalyzátory deaktivují.[3]
Reaktivita enů
I když jsou sterické efekty stále významné při určení výsledku enových reakcí katalyzovaných Lewisovými kyselinami, tak mají významný vliv na reaktivitu i elektronové jevy, protože v těchto reakcích vzbiká na centrálním uhlíkovém atomu enu významný kladný náboj. V důsledku toho jsou alkeny s alespoň jedním disubstituovaným vinylovým uhlíkem mnohem reaktiuvnější než mono- nebo 1,2-disubstituované alkeny.[3]
Mechanismus
Enové reakce katalyzovaných Lewisovými kyselinami mohou proběhnout soustředěným mechanismes, při kterém se tvoří polární meziprodukt, nebo postupným mechanismem s amfiontovým meziproduktem. Energie těchto mechanismů ovlivňuje výběr enu, enofilu i katalyzátoru; obecně platí, že čím reaktivnější je en nebo komplex enofilu a Lewisovy kyseliny, tím více je upřednostňován postupný mechanismus.[3]

Chirální Lewisovy kyseliny používané při asymetrických enových reakcích
Chirální dialkoxytitanové komplexy a syntéza laulimalidu
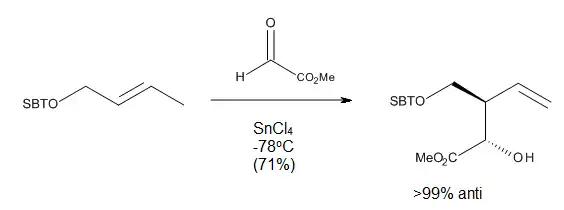
K aktuálně studovaným tématům v oblasti enových reakcí katalyzovaných Lewisovými kyselinami patří vývoj asymetrické tvorby vazeb C-C. Bylo popsáno použití chirálního komplexu titanu (viz následující obrázek) při asymetrické enové reakci za vzniku prochirálního glyoxylátového esteru.[14] Katalyzátor byl připaven na místě z (i-PrO)2TiX2 a opticky čistého binaftolu, výměna alkoxyligandů byla provedena pomocí molekulárních sít. Reakcí vznikají α hydroxyestery s vysokou enantiomerní čistotou, látky, které jsou významné biologicky i synteticky.[14]
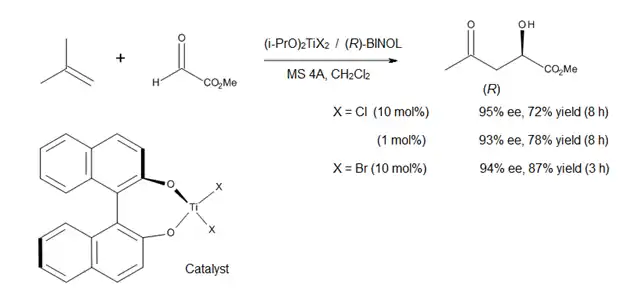
Jelikož jsou (S)-BINOL i (R)-BINOL běžně dostupné v enantiomerně čisté podobě, tak lze tímto postupem připravit oba enantiomery α hydroxyesterů a jejich derivátů. Uvedenou reakci lze ovšem použít pouze na 1,1-disubstituované alkeny, protože komplex titan-BINOL nemá příliš velkou kyselost.[14]
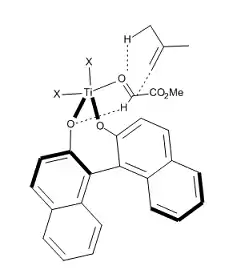
Roku 1997 byl navrhnut počáteční meziprodukt této reakce[15] za účelem vysvětlení pozorované vysoké enantioselektivity (za předpokladu, že je reakce exotermní, jak bylo vypočítáno ze standardních vazebných energií). I když struktura aktivního katalyzátoru není známa, tak tento model předpovídá následující: aldehyd je aktivován komplexací s chirálním katalyzátorem (R)-BINOL-TiX2 s formylovým volným elektronovým párem v poloze syn vzhledem k formylovému vodíku za vzniku struktuchiálně katalyzovanoury s koordinačním číslem 5. Vazba CH—O se opbjevuje na stereoelektronově nejvíce upřednostňovaném volném elektronovém páru kyslíku ligandu. V této látce je vrchní strana formylové skupiny mnohem lépe dostupná pro nukleofilní atak, protože spodní strana je krytá naftolovou skupinou.
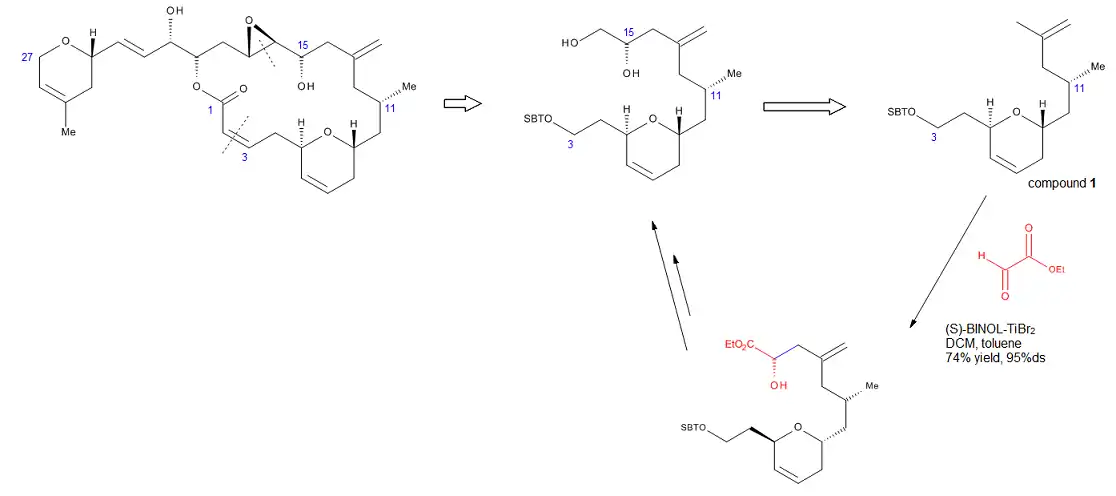
Roku 2002 byla popsána totální syntéza laulimalidu,[16] s využitím enové reakce - C3-C16 fragment jeho molekuly se vytváří chirálně katalyzovanou enovou reakcí vytvářející C15 stereocentrum. Reakcí koncové allylové skupiny sloučeniny 1 s ethylglyoxylátem za přítomnosti (S)-BINOL-TiBr2 jako katalyzátoru vzniká požadovaný alkohol se 74% výtěžností a 95% diastereomerním přebytkem.
Při tomto postupu není třeba připojovat na konec molekuly chránicí skupinu. Provedením této reakce se lze vyhnout tvrdším podmínkám a nižším výtěžkům spojeným se zaváděním exomethylenových skupin v dalším půběhu syntézy.[16]
C2-symetrické měďnaté komplexy a syntéza (+)-azaspiracidu-1
D. A. Evans se svými apolupracovníky[17] nový druh enantioselektivních C2-symetrických měďnatých katalyzátorů, které mohou zajišťovat chelaci substrátů prostřednictvím dvou karbonylových skupin. Tyto katalyzátory mají vysokou míru asymetrické indukce při některých reakcích, jako jsou enové reakce ethylglyoxylátu s různými nenasycenými alkeny.


U prvních dvou katalyzátorů se asymetrickou indukcí vytváří čtvercový komplex katalyzátoru s glyoxylátem, jak je zobrazeno na následujícím obrázku, přičemž je Re strana aldehydu stíněna terc-butylovými substituenty, díky čemuž alkeny atakují pouze Si stranu.[18]
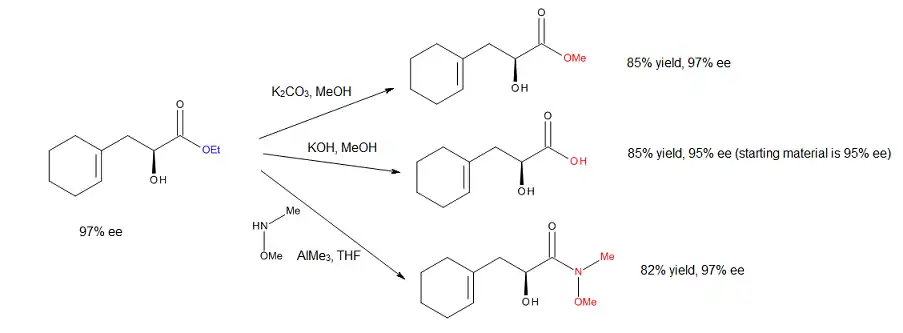
Význam postupu vyvinutého Evansem a spolupracovníky byl potvrzen úspěšnou přeměnou vznikajícího alfa-hydroxyesteru na odpovídající methylester, volnou kyselinu, Weinrebův amid a alfaazidoester, a to bez jakékoliv racemizace.[17] Azidové odstranění alkoholu představuje užitečný způsob přípravy ortogonálně chráněných aminokyselin.
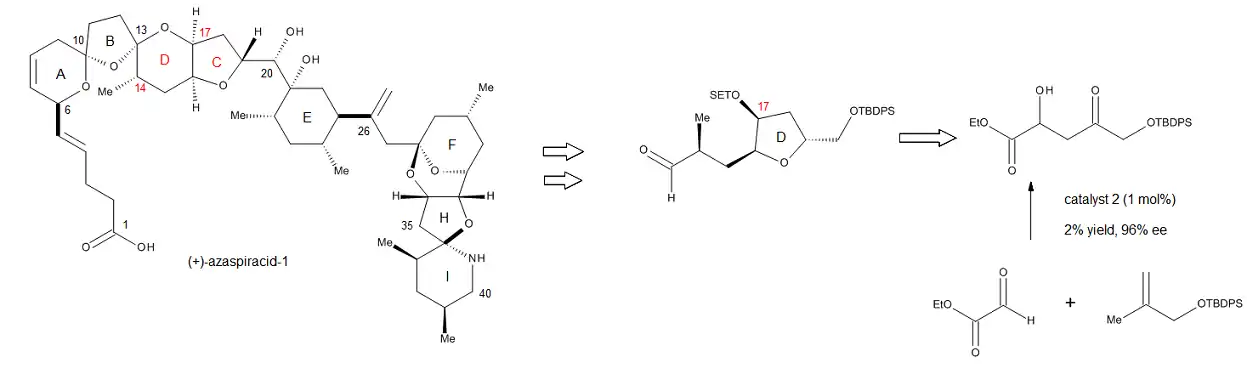
Syntetická přínosnost C2-symetrické měďnatých katalyzátorůl byla potvrzena přípravou C17 stereocentra části (+)-azaspiracid-1 kruhu (+)-azaspiracidu-1, což je jed vytvářený živočichy jako jsou ústřice, hřebenatky, škeble a srdcovky.[19] Reakce vytvářející toto stereocentrum je katalyzována 1 molárním procentem měďnatého komplexu; autoři naznačují, že i při použití 20 g bude mít reakce velmi dobrý výtěžek a vysokou enantioselektivitu. Produkt je také možné, aniž by došlo ke ztrátě selektivity, převést na odpovídající Weinrebův amid, což umožňuje snadné zavedení C14 skupiny. Tento katalyytický enantioselektivní postup lze jednoduše zahrnout do složitějších syntéz, obzvláště na žačátku procesů, kdy je důležité dosáhnout vysoké výtěžnosti.
Reference
V tomto článku byl použit překlad textu z článku Ene reaction na anglické Wikipedii.
- K. Alder; F. Pascher; A. Schmitz. Über die Anlagerung von Maleinsäure-anhydrid und Azodicarbonsäure-ester an einfach ungesättigte Koh an einfach ungesättigte Kohlenwasserstoffe. Zur Kenntnis von Substitutionsvorgängen in der Allyl-Stellung. Chemische Berichte. S. 2.
- K. Mikami; M. Shimizu. Asymmetric ene reactions in organic synthesis. Chemical Reviews. 1992, s. 1021.
- B. B. Snider. Lewis-acid catalyzed ene reactions. Accounts of Chemical Research. 1980, s. 426.
- G. D. Paderes; W. L. Jorgensen. Computer-assisted mechanistic evaluation of organic reactions. 20. Ene and retro-ene chemistry. The Journal of Organic Chemistry. 1992, s. 1904.
- S. Inagaki; H. Fujimoto; K. J. Fukui. Orbital interaction in three systems. Journal of the American Chemical Society. 1976, s. 4693.
- I. Fernandez; F. M. Bickelhaupt. Alder-ene reaction: Aromaticity and activation-strain analysis. Journal of Computational Chemistry. 2012, s. 509–516. PMID 22144106.
- L. M. Stephenson; D. L. Bickelhaupt. Stereochemistry of an ene reaction of dimethyl azodicarboxylate. The Journal of Organic Chemistry. 1976, s. 3614.
- R. J. Loncharich; K. N. Houk. Transition structures of ene reactions of ethylene and formaldehyde with propene. The Journal of Organic Chemistry. 1987, s. 6947.
- Christoph Schnabel; Katja Sterz; Henrik MüLler; Julia Rehbein; Michael Wiese; Martin Hiersemann. Total Synthesis of Natural and Non-Natural Δ5,6Δ12,13-Jatrophane Diterpenes and Their Evaluation as MDR Modulators. The Journal of Organic Chemistry. 2011, s. 512. PMID 21192665.
- MIKAMI, K.; SHIMIZU, M. Asymmetric ene reactions in organic synthesis. Chem. Rev.. 1992, s. 1021. DOI 10.1021/cr00013a014. (anglicky)
- H. M. R. Hoffmann. The Ene Reaction. Angewandte Chemie International Edition. 1969, s. 556.
- W. A. Thaler; B. J. Franzus. The Reaction of Ethyl Azodicarboxylate with Monoolefins. The Journal of Organic Chemistry. 1964, s. 2226.
- W. Oppolzer; V. Snieckus. Intramolecular Ene Reactions in Organic Synthesis. Angewandte Chemie International Edition in English. 1978, s. 476.
- K. Mikami; M. Terada; N. Takeshi. Catalytic asymmetric glyoxylate-ene reaction: A practical access to .alpha.-hydroxy esters in high enantiomeric purities. Journal of the American Chemical Society. 1990, s. 3949.
- E. J. Corey; D. Barnes-Seeman; T. W. Lee; S. N. Goodman. A transition-state model for the mikami enantioselective ene reaction. Tetrahedron Letters. 1997, s. 6513.
- M. R. Pitts; J. Mulzer. A chirally catalysed ene reaction in a novel formal total synthesis of the antitumor agent laulimalide. Tetrahedron Letters. 2002, s. 8471.
- D. A. Evans; S. W. Mulzer; C. S. Burgey; N. A. Paras; T. Vojkovsky. C2-Symmetric Copper(II) Complexes as Chiral Lewis Acids. Catalytic Enantioselective Carbonyl−Ene Reactions with Glyoxylate and Pyruvate Esters. Journal of the American Chemical Society. 2000, s. 7936.
- J. S. Johnson; D. A. Evans. Chiral bis(oxazoline) copper(II) complexes: Versatile catalysts for enantioselective cycloaddition, Aldol, Michael, and carbonyl ene reactions. Accounts of Chemical Research. 2000, s. 325–335. PMID 10891050.
- D. A. Evans; L. Kaerno; T. B. Dunn; Beauchemin; B. Raymer; J. A. Mulder; E. J. Olhava. Total synthesis of (+)-azaspiracid-1. An exhibition of the intricacies of complex molecule synthesis. Journal of the American Chemical Society. 2008, s. 16295–16309. PMID 19006391.