Needlemanův–Wunschův algoritmus
Needlemanův-Wunschův algoritmus je jedním ze základních algoritmů bioinformatiky. Provádí globální zarovnání sekvencí (tedy takové, ve kterém je v každé části zarovnání brán ohled na celou sekvenci) dovolující mezery, nejčastěji jde o sekvence nukleotidů v nukleových kyselinách či sekvence aminokyselin v proteinech. Jde o algoritmus dynamického programování, je to první algoritmus tohoto druhu použitý pro srovnávání biologických sekvencí. Byl sestaven v roce 1970 Saulem B. Needlemanem a Christianem D. Wunschem.
Vlastnosti
Algoritmus garantuje nalezení optimálního globálního zarovnání vzhledem k parametrům (tedy skórovací matici a sankcím za vložení mezery, viz dále). Prostorová složitost je kde a jsou délky zarovnávaných sekvencí. Původní algoritmus měl kubickou časovou složitost (a implicitně nepoužíval sankci za vložení mezery). Dnešní implementace mají kvadratickou časovou složitost a sankci za vložení mezery používají.



Princip
Vstupem algoritmu jsou 2 zarovnávané sekvence, skórovací matice a hodnota sankce za vložení mezery. Tradičně jde o celočíselné hodnoty. Ve skórovací matici jsou zapsány hodnoty skóre za shodu/neshodu příslušných znaků z abecedy používané v sekvenci. Algoritmus nejprve sestaví matici kde a jsou délky zarovnávaných sekvencí. Řádky matice přísluší jednotlivým znakům jedné sekvence, sloupce znakům druhé sekvence. První řádek a první sloupec nepřísluší žádnému znaku.

Postup:
= hodnota pole v matici na souřadnicích


= sankce za vložení mezery (záporné číslo nebo 0)

= hodnota skóre za shodu/neshodu znaků příslušejících poli v matici na souřadnicích podle skórovací matice

- Do levého horního rohu matice zapíšeme hodnotu skóre 0.
- Do každého dalšího pole matice zapíšeme hodnotu (tedy nejvyšší skóre, jakého lze v tomto poli dosáhnout. Bude to buď skóre pole vlevo + sankce za vložení mezery (inzerce) nebo skóre pole nahoře + sankce za vložení mezery (delece) nebo skóre pole vlevo nahoře + hodnota za shodu/neshodu znaků na pozicích příslušných tomuto poli podle skórovací matice) a zapamatujeme si, která z těchto tří možností byla oním maximem (tedy z kterého pole jsme se do tohoto dostali, při shodě si zapamatujeme všechny).
- Po zapsání takovýchto hodnot do všech polí matice začneme od pravého dolního pole (které do nejlepšího zarovnání určitě patří a zároveň je v něm zapsáno celkové skóre tohoto zarovnání) a pozpátku postupně zjišťujeme, ze kterých polí jsme se do tohoto a každého z jeho předchůdců dostali. Výsledná cesta v matici reprezentuje optimální zarovnání.

Příklad
Zarovnáváme nukleotidové sekvence ATCGAC a CATAC. Sankce za vložení mezery je . Skórovací matice vypadá takto:


(tedy hodnota za shodu/neshodu je pro všechny možnosti stejná. To nemusí platit vždy, především pro sekvence aminokyselin, kde se pravděpodobnosti záměny mezi jednotlivými aminokyselinami liší)
Matice po dokončení algoritmu vypadá takto: (modré šipky naznačují předchůdce jednotlivých polí, červenými šipkami je vyznačena cesta optimálního zarovnání maticí)
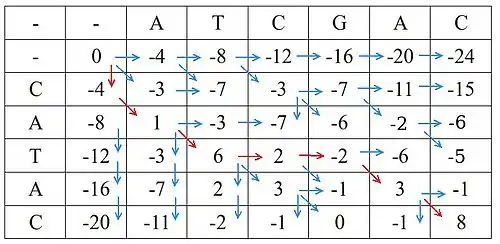
Výsledné zarovnání je tedy toto:
ATCGAC || || CAT--AC
Skóre nejlepšího zarovnání je 8.
Použití
Algoritmus je vhodný na zarovnávání kratších sekvencí spíše větší podobnosti a podobné délky. Na méně podobné či různě dlouhé sekvence je lepší lokální zarovnání algoritmem Smith-Waterman. Pro srovnávání dlouhých sekvencí na úrovni částí chromozomu jsou algoritmy dynamického programování nepoužitelné, kvůli příliš velké časové a prostorové náročnosti. V těchto případech se používají heuristické algoritmy jako BLAST, BLAT či PatternHunter, které nezaručují optimální zarovnání.
Je vidět, že matice obsahuje skóre všech možných zarovnání daných sekvencí. Proto může být použita pro srovnávání a hodnocení jiných (heuristických) algoritmů, které nemusí najít optimální řešení.