BLAST
BLAST (Basic Local Alignment Search Tool) je algoritmus používaný v bioinformatice za účelem srovnávání primárních sekvenčních informací, například nukleotidů DNA z různých sekvencí nebo sekvencí aminokyselin z různých proteinů. BLAST umožňuje srovnání dotazované (zadávané) sekvence se sekvencemi v databázi a zároveň rozpoznání obdobných sekvencí nad definovanou hranicí podobnosti. BLAST jako program navrhli Stephen Altschul, Warren Gish, Webb Miller, Eugene Myers, and David J. Lipman z NIH (National Institutes of Health) v USA a publikovali v Journal of Molecular Biology v roce 1990.[1]
Algoritmus
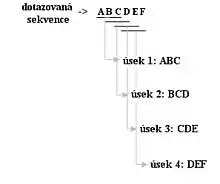
BLAST pro svůj průběh vyžaduje dotazovanou sekvenci (kterou hledáme) a cílovou sekvenci nebo sekvenční databázi (ve které hledáme, proti které hledáme). BLAST hledá subsekvence v databázi, které jsou podobné subsekvenci dotazované, přičemž v typickém případě je dotazovaná sekvence mnohem menší než databáze. Jednou z hlavních myšlenek BLASTu je, že statisticky signifikantní alignment obsahuje tzv. high-scoring segment páry (HSP). Tyto alignmenty (přiřazení) mezi dotazovanou sekvencí a sekvencemi v databázi vyhledává pomocí heuristických přístupů blízkým přesnějšímu algoritmu Smith-Waterman, který je ovšem pro prohledávání velkých genomových databází příliš pomalý. Více než padesátkrát rychlejší heuristický přístup dává spolu se stále ještě relativně dobrou přesností programům založených na BLAST velkou výhodu.[2]
Průběh algoritmu
BLAST provádí sekvenční alignment v několika krocích. Nejdříve jsou z dotazované sekvence odfiltrovány low-complexity regions a repetice, které by mohly negativně ovlivnit nalezení signifikantních sekvencí v databázi. Následně vzniká k-písmenný seznam úseků z dotazované sekvence, kdy k označuje délku úseku a je obvykle pro proteinové sekvence k=3, pro DNA sekvence k=11. Tyto úseky jsou pak postupně přiřazovány, jak je vidět na obr. 1. Tento seznam tedy obsahuje všechny možné varianty úseků dotazované sekvence a tyto úseky následně hledáme v databázi. Zda úsek odpovídá databázi je hodnoceno pomocí skórující (substituční) matrix, která přiřazuje skóre ("body") pro jednotlivé kombinace v páru, přičemž je úsek hodnocen jako odpovídající, pokud přesáhne předem definovanou prahovou hodnotu (threshold). Nejlépe odpovídající úseky jsou pak označovány jako high-scoring páry (HSP). V dalším kroku je pak alignment za užití stále stejné skórující matrix rozšířen na obě strany až do té doby, než skóre přestane dosahovat prahové hodnoty, tedy dokud celkový součet skóre high-scoring párů HSP nezačne klesat. Jedním z následných vylepšení BLASTu je pak možnost alignmentu zahrnujícího mezery (gaps), kde je úsek s nejvyšším skóre oboustranně rozšiřován pomocí metod dynamického programování[3]
BLAST vs FASTA
BLAST i FASTA algoritmy v bioinformatice používají metodu heuristického hledání pro rychlý párový sekvenční alignment, hledající krátké úseky identických nebo vysoce podobných záznamů ve dvou sekvencích. Není zaručeno nalezení nejlepšího (optimálního) alignmentu nebo skutečných homologů, ovšem metody jsou 50×-100× rychlejší než dynamické programování. BLAST je díky hledání pouze vysoce signifikantních záznamů v sekvencích časově efektivnější než FASTA, přičemž citlivost je srovnatelná.[3]
Program
BLAST program je možné spustit dvěma způsoby a sice, stáhnout jako "blastall" a spustit jako nástroj příkazového řádku nebo používat přístupem přes web. Webový server BLASTu provozovaný NCBI umožňuje komukoliv s webovým prohlížečem prohledávat databáze, které obsahují většinu osekvenovaných organismů. BLAST program je založen jako open-source, čímž dává možnost modifikace programového kódu, díky čemuž vzniklo několik BLASTových vylepšení. BLAST je skupina programů, které jsou používány v závislosti na cíli hledání a vstupních datech. Tyto verze se liší v zadávaných vstupních datech, prohledávaných databázích a ve vlastním srovnávání.[4]
Nukleotid-nukleotid BLAST (blastn)
Program vrací k zadané dotazované sekvenci DNA nejvíce podobné sekvence DNA z databáze DNA vybrané uživatelem.
Protein-protein BLAST (blastp)
Program vrací k zadané dotazované proteinové sekvenci nejvíce podobné proteinové sekence z databáze proteinů vybrané uživatelem.
Position-Specific Iterative BLAST (PSI-BLAST, blastpgp)
Program používaný k hledání vzdáleně příbuzných proteinů. Nejdříve je vytvořen seznam blízce příbuzných proteinů, které jsou následně zkombinovány jako obecná "profilová" sekvence shrnující signifikantní znaky těchto sekvencí. Tato sekvence je poté srovnávána s databází, přičemž je nalezeno velké množství proteinů. Tyto proteiny jsou opět použity pro vytvoření "profilové" sekvence a celý proces se opakuje. Při hledání proteinů má PSI-BLAST vyšší citlivost zachycení příbuzných proteinů, neboť je schopen najít i vzdálené evoluční vztahy.
Nucleotide 6-frame translation-protein (blastx)
Program porovnává 6-rámcové abstraktní translační produkty dotazované sekvence nukleotidů (obě vlákna) s proteinovou sekvenční databází.
Nucleotide 6-frame translation-nucleotide 6-frame translation (tblastx)
Nejpomalejší z programů BLASTu překládá dotazovanou nukleotidovou sekvenci do všech šesti možných čtecích rámců a srovnává ji s 6 rámcovou translací v nukleotidové sekvenční databázi. TBLASTX se takto pokouší nalézt velmi vzdálené vztahy mezi nukleotidovými sekvencemi.
Protein-nucleotide 6-frame translation (tblastn)
Program porovnává dotazovanou sekvenci proteinu se všemi šesti čtecími rámci v databázi sekvence nukleotidů.
Velký počet dotazovaných sekvencí – Large numbers of query sequences (megablast) Při porovnávání velkého počtu dotazovaných sekvencí pomocí BLASTu přes příkazový řádek je "megablast" mnohem rychlejší než několikanásobné spuštění BLASTu. Megablast zřetězí několik vstupních sekvencí dohromady ještě před prohledáváním databáze a následně analyzuje výsledky vyhledávání pro získání jednotlivých alignmentů a statistických hodnot.
Užití BLASTu
BLAST je možné využít pro řadu procesů mezi něž patří například identifikace druhů, lokalizace domén, stanovování fylogeneze a DNA mapování či porovnávání.
Identifikace druhů
S použitím BLASTu je možná identifikace pravděpodobnosti identity druhu a/nebo nalezení druhů homologních, čehož se využívá například při práci s DNA neznámých druhů.
Lokalizace domén
Možnost lokalizace známých domén v sekvenci proteinu.
Stanovení fylogeneze
Za pomoci BLAST web-stránky je možné sestavit z výsledků BLASTu fylogenetický strom. Ovšem fylogeneze založená pouze na BLAST samotném je méně spolehlivá než další výpočetní fylogenetické metody, je proto vhodná pouze pro prvotní analýzu.
DNA mapování
Při práci se známými druhy, kdy dotazujeme sekvenci genu s neznámou lokalizací může BLAST porovnat pozice dotazovaných sekvencí na chromozomu s relevantními sekvencemi v databázi (databázích).
Srovnávání
Při práci s geny může BLAST lokalizovat společné geny ve dvou příbuzných druzích, což může být použito pro mapování anotací z jednoho organismu pro další.
Reference
V tomto článku byl použit překlad textu z článku BLAST na anglické Wikipedii.
- ALTSCHUL, S; GISH, W; MILLER, W; MYERS, E; LIPMAN, D. Basic local alignment search tool. Journal of Molecular Biology. 1990, s. 403–410. Dostupné online. DOI 10.1016/S0022-2836(05)80360-2. PMID 2231712. (anglicky)
- MOUNT, D. W. Bioinformatics: Sequence and Genome Analysis. 2nd. vyd. [s.l.]: Cold Spring Harbor Press, 2004. Dostupné online. ISBN 978-0-8796-9712-9. (anglicky)
- Xiong, J. Essential Bioinformatics. [s.l.]: Cambridge University Press, 2006. ISBN 978-0-521-60082-8. (anglicky)
- Program Selection Tables of the Blast NCBI web site [online]. Dostupné online. (anglicky)
| Autoritní data |
|
|---|