Pompeho choroba
Pompeho choroba (též glykogenóza typu II, GSD II, deficit kyselé maltázy) je vzácná choroba a autozomálně recesivní metabolická porucha, která poškozuje svalové a nervové buňky. Je způsobena mutací genu pro lysozomální kyselou α-1,4-glukosidázu (GAA neboli kyselou maltázu).[1] Následkem deficitu nebo nedostatečné aktivity enzymu GAA dochází k akumulaci lysozomálního glykogenu v mnoha tkáních, především v kosterních svalech a u kojenců i v myokardu, v menší míře také v endotelu cévního systému, v centrální nervové soustavě (CNS) především v astrocytech, v játrech a ledvinách.[1] To způsobuje progresivní svalovou slabost (myopatii), která postihuje různé tělní tkáně.
| Pompeho choroba | |
|---|---|
| Klasifikace | |
| MKN-10 | E74.0 |
| MeSH | D006009 |
| Statistické údaje – obě pohlaví[1] | |
| Incidence | 1:40 000 |
| Některá data mohou pocházet z datové položky. | |
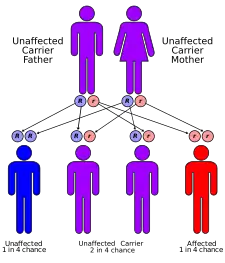
Rozlišují se dva typy onemocnění, a to infantilní (postihuje novorozence) a juvenilní + adultní (postihuje starší děti a dospělé). Nemoc je pojmenována podle nizozemského patologa J. C. Pompeho, který ji v roce 1932 popsal.[1]
Odkazy
Reference
V tomto článku byl použit překlad textu z článku Glycogen storage disease type II na anglické Wikipedii.
- V tomto článku je použit text článku Glykogenózy ve WikiSkriptech českých a slovenských lékařských fakult zapojených v MEFANETu.
- SLOUKOVÁ, Eva; OŠLEJŠKOVÁ, Hana; VOHÁŇKA, Stanislav; JEŠINA, Pavel. Pompeho choroba. Pediatrie pro praxi. 2009, roč. 10, čís. 3, s. 156–158. Dostupné online.
Externí odkazy
 Obrázky, zvuky či videa k tématu Pompeho choroba na Wikimedia Commons
Obrázky, zvuky či videa k tématu Pompeho choroba na Wikimedia Commons - Pompeho choroba v databázi Who Named It? (anglicky)
- (česky) Česká televize – Medicína pro 21. století: Pompeho nemoc
- (česky) Sdružení Meta – Pompeho choroba
Přečtěte si prosím pokyny pro využití článků o zdravotnictví.