Oxy-Copeův přesmyk
Oxy-Copeův přesmyk je organická reakce spočívající v přeměně struktury některých nenasycených alkoholů; jedná se o variantu Copeova přesmyku, v níž se 1,5-dien-3-oly přeměňují na nenasycené karbonylové sloučeniny mechanismem odpovídajícím [3,3]-sigmatropním přesmykům.[1][2]
Tato reakce je předvídatelná a snadno proveditelná u řady různých substrátů, díky čemuž je i velmi často používaná.[3]
Další výhodou je často jednoduchá příprava výchozích látek. Hlavní částí je tvorba karbonylové skupiny keto-enol tautomerizací.[4]
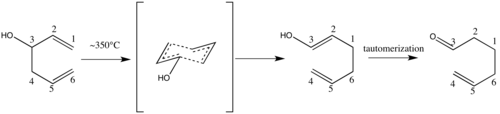
S použitím zásady probíhá reakce 1010-1017krát rychleji; tato podoba se nazývá aniontový oxy-Copeův přesmyk.[5]
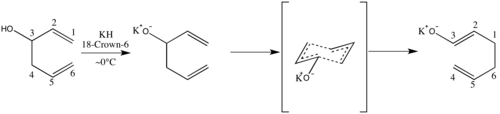
Tvorba enolátu způsobuje, že je reakce většinou nevratná.[3][4][6]
Historie
Sigmatropní přesmyky jsou významným nástrojem v organické syntéze.[6] K předvedení využitelnosti Copeova přesmyku byl použit alkohol s hydroxylovou skupinou v poloze C-3 1,5-dienu, zahříváním plynného bicyklického dienolu v plynném skupenství byl získán cis-∆5,6-oktalon.[1]
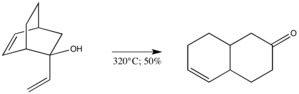
Oxy varianta umožňuje některé postupy, které byly dříve neproveditelné.
V roce 1975 bylo popsáno výrazné urychlení reakce při přidání zásad, jako je hydrid draselný, které se poté stalo běžným. V některých případech však přítomnost aniontů není kvůli přílišné citlivosti vznikajícího enolátu žádoucí; příkladem je následující reakce, z níž byl získán pouze dehet.[7]
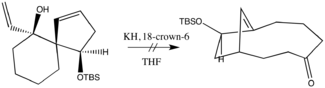
Původní oxy-Copeův přesmyk je tak stále významnou součástí syntetické chemie.
Mechanismus
Jak neutrální, tak i aniontové oxy-Copeovy přesmyky mohou probíhat jedním ze dvou mechanismů: soustředěným nebo postupným radikálovým, i když většinou převládá první z nich.[8][9] Meziprodukt převažujícího mechanismu zaujímá židličkovou konformaci.[10]
Přesuny chirálních skupin jsou usnadněny vysoce uspořádanými přechodnými stavy.[4][10] Stereochemii produktu určuje poloha dvojné vazby u nejsnáze vznikajícího přechodného stavu.[3] Lodičková konformace je nevýhodná, ovšem existují i přesmyky s významným podílem této varianty, jenž vedou k tvorbě směsí diastereomerů.
![]()
Na reakci mohou mít velký vliv sterické efekty.[11]
Probíhat mohou i přesmyky, u kterých se židličkovité meziprodukty z geometrických důvodů nemohou utvořit. Vznik enolátu je dostatečně silným hnacím mechanismem k překonání energetické bariéry spojené s dearomatizací i lodičkovou konformací.[12]
Soustředěné mechanismy převládají; bylo vypočteno, že u aniontového oxy-Copeova přesmyku je heterolýza energeticky o 70-140 kJ/mol výhodnější než homolýza.[13]
Existuje několik faktorů, které mohou překonat tuto energetickou překážku.[10]
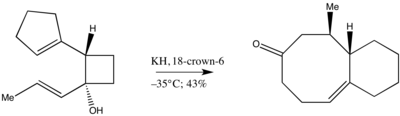
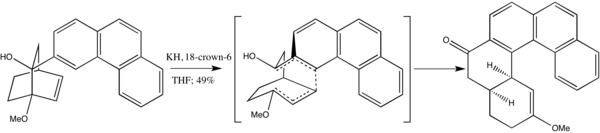
Kvůli velkému sterickému napětí způsobovanému methylovými skupinami při přípravě cyklooktenonu převažuje (Z)-izomer nad (E)-izomerem, což naznačuje, že meziprodukt se netvoří současně. Vznik pozorovaného produktu lze vysvětlit fragmentací a následnou izomerizací.[10]
Ve studii zaměřené na aniontové oxy-Copeovy přesmyky v plynné fázi se ukázalo, že urychlení reakce není způsobováno interakcemi s rozpouštědlem, ale samotnou strukturou.[14]
Urychlení reakce
Obecně platí, že snížení stability oxy-Copeova aniontového či oxy-Copeova substrátu oproti produktu vede k rychlejšímu průběhu reakce; lze toho dosáhnout několika různými způsoby. Reakci významně ovlivňují iontové interakce mezi kovem a alkoxidem, kdy disociativní vlastnosti vyvolávají navýšení rychlosti.[5] Použití 15-crown-5 s hydridem sodným vedlo k o 27 % rychlejší reakci než sigmatropní přesmyk alkoxidu bicyklického dienu na enolát, zatímco při použití hexamethylfosforamidu (HMPT) nebyl zaznamenán žádný vliv na rychlost reakce. Podobný účinek, s přibližně 180násobným urychlením, mělo použití hydridu draselného s 18-crown-6.
Použití polárnějších rozpouštědel a katalytického množství katalyzátorů fázového přenosu mělo podobné účinky.[15]
Průběh reakce usnadní, a tedy i urychlí, rovněž omezení kruhového napětí u reaktantů.
Využití
Jelikož existuje mnoho přírodních látek obsahujících osmičlenné cykly, jejichž syntéza se ukázala jako obtížná, tak je vhodným náhradním způsobem jejich přípravy aniontový oxy-Copeův přesmyk. Jeho použití umožňuje řízení stereochemie produktů.[16]
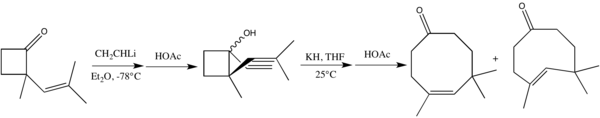
I přes možné geometrické vlivy mohou použité nenasycené substráty obsahovat také trojné vazby. Příslušné alkynoly byly využity například při syntéze poitediolu a daktylolu.[6] Takovéto sigmatropní přesmyky mohou probíhat aniontově i tepelně.[17]
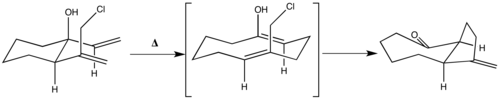
Předmětem zájmu je také využití oxy-Copeových přesmyků tam, kde jejich produkt dále reaguje za vzniku konečného produktu. Příkladem je níže zobrazená příprava cis-hydroazulenonu, u níž stereoelektronová konfigurace oxy-Copeova meziproduktu dovoluje SN odštěpení.[18]
Další okolnosti
Hydrid draselný, reaktant často používaný v aniontových oxy-Copeových přesmycích, někdy obsahuje stopová množství nečistot, které mohou poškodit dienolátový meziprodukt a vyvolat tak polymerizaci. Před provedením reakce se doporučuje přidání jodu, který odstraní případné pozůstatky superoxidu draselného. Tímto postupem lze výrazně vylepšit výtěžnost reakce i opakovatelnost výsledků.[19]
K vedlejším reakcím patří heterolytické štěpení, při němž se homoallylové alkoholy rozkládají na karbonylovou a allylovou sloučeninu.[20]
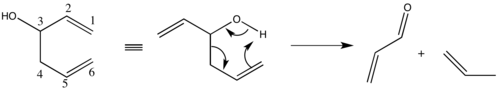
Tyto vedlejší reakce je možné potlačit omezením iontové povahy vazby kov-alkoxid, konkrétně použitím elektronegativnějších alkalických kovů nebo rozpouštědel méně náchylných k solvataci kationtů.[21]
Reference
V tomto článku byl použit překlad textu z článku Oxy-Cope rearrangement na anglické Wikipedii.
- Jerome A. Berson; Maitland Jones. A Synthesis of Ketones by the Thermal Isomerization of 3-Hydroxy-1,5-hexadienes. The Oxy-Cope Rearrangement. Journal of the American Chemical Society. 1964, s. 5019–5020. DOI 10.1021/ja01076a067.
- Arthur C. Cope; Elizabeth M. Hardy. The Introduction of Substituted Vinyl Groups. V. A Rearrangement Involving the Migration of an Allyl Group in a Three-Carbon System. Journal of the American Chemical Society. 1940, s. 441–444. DOI 10.1021/ja01859a055.
- Leo A. Paquette. Recent Applications of Anionic Oxy-Cope Rearrangements. Tetrahedron. 1997, s. 13971–14020. DOI 10.1016/S0040-4020(97)00679-0.
- Lázló Kürti; Barbara Czakó. Strategic Applications of Named Reactions in Organic Synthesis.Chybí název periodika! Burlington: Elsevier, 2005, s. 325.
- D. A. Evans; A. M. Golob. [3,3]Sigmatropic Rearrangements of 1,5-Diene Alkoxides. The Powerful Accelerating Effects of the Alkoxide Substituent. Journal of the American Chemical Society. 1975, s. 4765–4766. DOI 10.1021/ja00849a054.
- Stephen R. Wilson. Anion-Assisted Sigmatropic Rearrangements. Organic Reactions. 1993, s. 93–250. ISBN 0471264180. DOI 10.1002/0471264180.or043.02.
- Leo A. Paquette; Gaetan Ladouceur. Synthetic Studies Targeted at the Cytotoxic 8,9-Seco-ent-kaurene Diterpenes. Concise Complementary Stereocontrolled Construction of the Bridgehead Olefin Core. The Journal of Organic Chemistry. 1989, s. 4278–4279. DOI 10.1021/jo00279a010.
- D. A. Evans; D. J. Baillargeon. Alkoxide Substituent Effects on Carbon—Carbon Bond Homolysis. Tetrahedron Letters. 1978, s. 3319–3322. DOI 10.1016/S0040-4039(01)85627-6.
- Leo A. Paquette; Francis Pierre; Charles E. Cottrell. Anionic Rearrangements of syn- and anti-7-Cyclopentenyl-7-hydroxynorbornenes. The Case for Sequential Ring Cleavage and Intramolecular Michael Addition. Journal of the American Chemical Society. 1987, s. 5731–5740. DOI 10.1021/ja00253a027.
- Philippe Maurin; Se-Ho Kim; Sung Yun Cho; Jin Kun Cha. On the Mechanism of the Anionic Oxy-Cope Rearrangement of trans-1,2-Dialkenylcyclobutanols. Angewandte Chemie. 2003, s. 5044–5047. DOI 10.1002/anie.200350988. PMID 14595626.
- D. A. Evans; John V. Nelson. Stereochemical Study of the [3,3] Sigmatropic Rearrangement of 1,5-Diene-3-alkoxides. Application to the Stereoselective Synthesis of (.+-.)-Juvabione. Journal of the American Chemical Society. 1980, s. 774–782. DOI 10.1021/ja00522a056.
- Yasushi Ogawa; Tetsuya Ueno; Michinori Karikomi; Katsura Seki; Kazuo Haga; Tadao Uyehara. Synthesis of 2-Acetoxy[5]helicene by Sequential Double Aromatic Oxy-Cope Rearrangement. Tetrahedron Letters. 2001, s. 7827–7829. DOI 10.1016/s0040-4039(02)01611-8.
- D. A. Evans; D. J. Baillargeon. Intrinsic Fragmentation Modes of Primary Alkoxides. Tetrahedron Letters. 1978, s. 3315–3318. DOI 10.1016/S0040-4039(01)85626-4.
- John E. Baldwin; Kersey A. Black. Complete Kinetic Analysis of Thermal Stereomutations among the Eight 2,3-Dideuterio-2-(methoxymethyl)spiro[cyclopropane-1,1'-indenes]. Journal of the American Chemical Society. 1984, s. 1029–1040. DOI 10.1021/ja00316a036.
- Michael Georges; Tim F. Tam; Bert Fraser-Reid. Controlled Access to Furanose Precursors Related to Sesquiterpene Lactones 1. The Journal of Organic Chemistry. 1985, s. 5747–5753. DOI 10.1021/jo00350a062.
- Robert C. Gadwood; Renee M. Lett. Preparation and Rearrangement of 1,2-Dialkenylcyclobutanols. A Useful Method for Synthesis of Substituted Cyclooctenones. The Journal of Organic Chemistry. 1982, s. 2268–2275. DOI 10.1021/jo00133a007.
- Alfred Viola; John H. MacMillan. Vapor Phase Acetylenic Oxy-Cope Reaction of 5-Hexen-1-yn-3-ol. The Chemistry of an Allenol Intermediate. Journal of the American Chemical Society. 1970, s. 2404–2410. DOI 10.1021/ja00711a034.
- Michael Sworin; Ko Chung Lin. Cyclopentanoid Synthesis via the Intramolecular Trapping of Oxy-Cope Intermediates. Stereocontrolled Synthesis of the cis- and trans-Hydroazulene Skeleton. The Journal of Organic Chemistry. 1987, s. 5640–5642. DOI 10.1021/jo00234a029.
- Timothy L. Macdonald; Kenneth J. Natalie; Girija Prasad; J. Scott Sawyer. Chemically Modified Potassium Hydride. Significant Improvement in Yields in Some Oxy-Cope Rearrangements. The Journal of Organic Chemistry. 1986, s. 1124–1126. DOI 10.1021/jo00357a035.
- Roger L. Snowden; Bernard L. Muller; Karl H. Schulte-Elte. Fragmentation of Homoallylic Alkoxides. Synthesis of Propenyl and 2-Methylpropenyl Ketones from Carboxylic Esters. Tetrahedron Letters. 1981, s. 335–338. DOI 10.1016/S0040-4039(00)86824-0.
- D. A. Evans; D. J. Baillargeon; John V. Nelson. A General Approach to the Synthesis of 1,6-Dicarbonyl Substrates. New Applications of Base-Accelerated Oxy-Cope Rearrangements. Journal of the American Chemical Society. 1978, s. 2242–2244. DOI 10.1021/ja00475a051.