Fylogenetický strom
Fylogenetický strom („strom života“) je grafické zobrazení připomínající strom, jímž se znázorňují příbuzenské vztahy mezi různými biologickými druhy či jinými taxonomickými jednotkami, o nichž se předpokládá, že mají jednoho společného předka. Každé větvení (uzel) představuje hypotetického posledního společného předka. Každá větev znamená jednu evoluční linii, na jejímž konci jsou dané taxony. Do fylogenetického stromu je možné také zaznamenávat genové toky (tj. výměnu genů a mezidruhová křížení) spojnicemi jednotlivých větví, čas (obvykle na vertikální ose) a vzájemnou vzdálenost druhů (anagenezi).
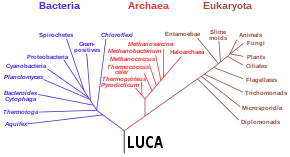
Rozdělování druhu do jednotlivých větví (tj. reprodukční izolace) se nazývá fylogeneze. Strom zaznamenávající nikoliv rodokmen, nýbrž fenotypickou podobnost organismů označujeme jako fenogram.[1]
Strom ovšem díky neúplnosti může odpovídat mnoha historiím diverzifikace druhů.[2]
Vytváření, manipulace a analýza fylogenetických stromů je možná např. v prostředí R za pomoci balíku ape či pokročilejšího balíčku Phytools.
Vytvoření fylogenetického stromu
V současnosti existuje několik možností analýzy příbuzenských vztahů mezi taxonomickými jednotkami různé úrovně. Podkladem pro vytvoření fylogenetického stromu je soubor dat, odrážející podobnosti a rozdíly mezi jedinci dané populace, například tabulka sekvencí nukleotidů nebo aminokyselin pro vybraný analyzovaný gen/protein. Ty jsou následně seřazeny do mnohonásobného porovnání sekvencí (MSA, z angl. multiple sequence alignment) pomocí algoritmů jako ClustalW, MAFFT nebo Muscle, jejichž principem je vytvoření matice, ve které jsou fylogeneticky příbuzné nukleotidy/aminokyseliny seřazeny na odpovídající pozici nad sebou. Data z tohoto souboru jsou následně využita pro výpočet vzdálenostní matice (z angl. distance matrix), shrnující teoretické vzdálenosti mezi jednotlivými vzorky souboru, ze které je pak příslušnou metodou vytvořen fylogenetický strom. Mezi nejčastěji využívané metody konstrukce fylogenetických stromů patří metody založené na hierarchickém shlukování (neighbor joining - NJ, UPGMA) nebo heuristické metody (maximální parsimonie, maximální věrohodnost), které navíc nevyžadují vzdálenostní matrici jako vstupní zdroj dat.
UPGMA
Tzv. metoda neváženého párování s aritmetickým průměrem (z angl. unweighted pair group method with arithmetic mean) je metoda konstrukce fylogenetických stromů založená na postupném vytváření párů z jedinců, mezi kterými je nejmenší fylogenetická vzdálenost, tj. jsou nejpodobnější. UPGMA probíhá na více úrovních (tzv. bottom-up), kdy v prvním kroku je z dvou nejvíce podobných jedinců určen počáteční pár. V dalším kroku je vypočtena vzdálenost mezi ostatními jedinci v populaci a tímto párem, přičemž ten nejméně vzdálený je shloučen do nového klastru obsahující původní klastr a nově přidaného jedince. Výpočet následně pokračuje až do určení všech klastrů. Po jejich určení je pak vytvořen fylogenetický strom, ve kterém distribuce jedinců mezi klastry slouží jako kvalitativní údaj (způsob větvení stromu) a vzdálenosti mezi jednotlivými klastry jako údaj kvantitativní (délky jednotlivých větví). Výslední strom je ultrametrický, což znamená že fylogenetické vzdálenosti k vybranému uzlu jsou mezi jedinci v rámci stejného páru identické.[3]
NJ
Podobně jako UPGMA, NJ je metoda vytváření fylogenetických stromů založena na principu hierarchického shlukování, prvně popsaná v roce 1987.[4] NJ vychází z populace jedinců, kteří jsou postupně shlukováni do klastrů na základě jejich vzájemné fylogenetické vzdálenosti, přičemž každý pár je tvořen nejméně vzdálenými (nejvíce podobnými) jedinci.[5] Na rozdíl od UPGMA ale NJ předpokládá variabilitu v evoluční rychlosti (např. rozdíly ve frekvenci mutací v čase) a proto je jejím výsledkem fylogenetický strom zahrnující odlišné vzdálenosti mezi jedinci tvořícím stejný pár a vybraným uzlem.
Maximální parsimonie
Maximální parsimonie je predikční metoda, která popisuje evoluci jedinců v rámci analyzovaného souboru pomocí stromu s nejnižším nutným počtem evolučních změn (např. nukleotidových změn mezi sekvencemi). Predikce je provedena z MSA (mnohonásobné porovnání sekvencí, z angl. multiple sequence alignment) studovaných sekvencí, kdy je pro každou variabilní pozici nalezen strom s nejmenším počtem změn vysvětlujícím její sekvenční variabilitu. Jako finální je vybrán tzv. maximálně parsimonní, tj. nejkratší strom. Jelikož maximální parsimonie vyžaduje nalezení co největšího počtu stromů k oskórování, při větším počtu srovnávaných jedinců (typicky nad 20) je nutné použít heuristické vyhledávání, kdy finální strom je určen ze souboru, který nezahrnuje všechny možné fylogenetické uspořádání. Další z nevýhod použití maximální parsimonie je fakt, že automaticky nalezený nejlépe skórující strom nemusí odpovídat skutečnému průběhu evoluce, ve které může docházet např. k reverzním mutacím.[6]
Maximální věrohodnost
Analogem maximální parsimonie při konstrukci fylogenetických stromů je metoda založená na odhadu tzv. maximální věrohodnosti (z angl. maximum likelihood). Principem této metody je průběžné vyhledávání a skórování fylogenetických stromů, přičemž jako výslední strom je použit ten, který nejlépe vysvětluje daná data na základě evolučního modelu. Pro metodu maximální věrohodnosti je nutný vstupný model (např. pravděpodobnost záměny jedné nukleotidové báze za jinou, pro každou možnou bázi), kterého použití zvyšuje šanci na nalezení stromu odpovídajícímu skutečnosti.[7] V porovnání s NJ, UPGMA nebo maximální parsimonií je analýza maximální věrohodnosti jako taká náročná na výpočetní techniku, zejména při vysokém počtu analyzovaných sekvencí, i přesto je ale v současnosti standardní metodou konstrukce fylogenetických stromů.
Metody založené na Bayesiánské inferenci
Mezi nejnovější metody konstrukce fylogenetických stromů patří metody založené na Bayesiánské inferenci (z angl. Bayesian inference). Tyto metody využívají předešlé znalosti o datech a vstupný model (podobně jako u maximální věrohodnosti) k nalezení fylogenetického stromu který nejlépe odpovídá datům (studovaným sekvencím). Hledání je započato vybraním náhodného stromu, jeho oskórováním a následným porovnáním s podobným fylogenetickým stromem; pro další analýzu se pak využije ten strom, který dosáhne vyšší pravděpodobnostní skóre. Tento postup je posléze opakován až do dosáhnutí rovnovážního stavu, tj. již nelze najít lépe skórující strom.[8] Jelikož jsou tyto metody výpočetné náročné, k jejich intenzivnějšímu využívání došlo až ke konci 20. století; v současnosti je již dostupná řada softwarů umožňujícím rutinní konstrukci fylogenetických stromů pomocí Bayesiánské inference jako např. MrBayes, BEAST, PhyloBayes nebo Bali-Phy.
Odkazy
Reference
V tomto článku byl použit překlad textu z článku Phylogenetic tree na anglické Wikipedii.
- FLEGR, J. Zamrzlá evoluce. [s.l.]: Academia, 2008.
- https://phys.org/news/2020-04-accuracy-methods-trees-life.html - Researchers challenge accuracy of methods that analyze trees of life
- SOKAL, R.E; MICHENER, C.D. A statistical method for evaluating systematic relationships. University of Kansas Science Bulletin. 1958, roč. 38, čís. 22, s. 1409–1438. Dostupné online.
- SAITOU, N.; NEI, M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Molecular Biology and Evolution. 1987-07, roč. 4, čís. 4, s. 406–425. PMID: 3447015. Dostupné online [cit. 2022-02-08]. ISSN 0737-4038. DOI 10.1093/oxfordjournals.molbev.a040454. PMID 3447015.
- GASCUEL, O.; STEEL, M. Neighbor-Joining Revealed. Molecular Biology and Evolution. 2006-11-01, roč. 23, čís. 11, s. 1997–2000. Dostupné online [cit. 2022-02-08]. ISSN 0737-4038. DOI 10.1093/molbev/msl072.
- MOUNT, D.W. Maximum Parsimony Method for Phylogenetic Prediction. Cold Spring Harbor Protocols. 2008-04, roč. 2008, čís. 4, s. pdb.top32. Dostupné online [cit. 2022-02-08]. ISSN 1940-3402. DOI 10.1101/pdb.top32. (anglicky)
- CHO, A. Constructing Phylogenetic Trees Using Maximum Likelihood. Scripps Senior Theses. 2012-04-09. Dostupné online [cit. 2022-02-08].
- NASCIMENTO, F.F.; DOS REIS, M.; YANG, Z. A biologist’s guide to Bayesian phylogenetic analysis. Nature Ecology & Evolution. 2017-10, roč. 1, čís. 10, s. 1446–1454. Dostupné online [cit. 2022-02-09]. ISSN 2397-334X. DOI 10.1038/s41559-017-0280-x. PMID 28983516. (anglicky)