Elektronová korelace
Elektronová korelace je interakce mezi elektrony v elektronové struktuře kvantového systému. Korelační energie je měřítkem toho, jak moc pohyb jednoho elektronu je ovlivněn přítomností všech ostatních elektronů.
Atomární a molekulární systémy
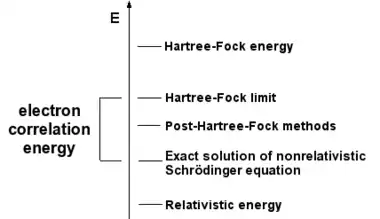
V rámci Hartreeho–Fockovy metody z kvantové chemie, je antisymetrická vlnová funkce aproximována pomocí jediného Slaterova determinant. Exaktní vlnové funkce, však nemůžou být obecně vyjádřeny jako jeden determinant. Jedno-determinantní přiblížení nebere v úvahu Coulombovskou korelaci, což vede k tomu, že celková elektronová energie se liší od přesného řešení nerelativistické Schrödingerovy rovnice v rámci Born–Oppenheimerovy aproximace. Proto, Hartreeho–Fockova limita je vždy nad touto exaktní energií. Rozdíl se nazývá korelační energie, termín vytvořený Löwdinem.[1] Pojem korelační energie byl už ale zkoumán dříve Wignerem.[2]
Určité množství elektronové korelace je již započítáno v rámci HF aproximace, konkrétně v elektronové výměně. Tento termín popisuje vztah mezi elektrony s paralelním spinem. Tato základní korelace zabraňuje dvěma elektronům s paralelním spinem, aby byly nalezeny na stejném místě v prostoru a je často nazývána Fermiho korelace nebo Fermiho díra. Coulombická korelace, na druhou stranu, popisuje korelaci mezi prostorovou pozici elektronů v důsledku jejich Coulombického odpuzování, a je zodpovědná za chemicky důležité efekty, jako je například Londonova disperze. Existuje také korelace související s celkovou symetrií nebo celkovým spinem uvažovaného systému.
Označení korelační energie musí být používána s opatrností. Jednou z obvyklých definic je, že je se jedná o energetický rozdíl korelované metody vzhledem k energii získané z Hartreeho–Fockovy metody.

Ale to není úplná korelační energie, protože jistá část korelace je již zahrnutá v HF. Za druhé, korelační energie je velmi závislá na použité bázi. „Exaktní“ energie je energie, s úplnou korelací a úplnou bází.
Elektronová korelace je někdy rozdělena na dynamickou a nedynamickou (statickou) korelaci. Dynamická korelace je korelace vycházející z pohybu elektronů a je zachycená např. v metodě konfigurační interakce (CI). Statická korelace je důležitá pro molekuly, kde je základní stav dobře popsán pouze s více než jedním (téměř) degenerovaným determinantem. V tomto případě je vlnová funkce v Hartreeho–Fockově metodě (pouze s jediným determinantem) kvalitativně špatná. Metoda multi-konfiguračního self-konzistentního pole (MCSCF) pokrývá statickou korelaci, ale ne dynamickou korelaci.
Metody
Jednoduše řečeno, molekulární orbitaly metody Hartree–Fock jsou optimalizovány vyhodnocením energie elektronu v každé molekulární orbitě jako pohyb v průměrném poli všech ostatních elektronů, spíše než zahrnující okamžitou repulzi mezi elektrony.
Pro získání elektronové korelace existuje mnoho post-Hartree–Fock metod, včetně:
Jednou z nejdůležitějších metod pro korekci chybějící korelace je metoda konfigurační interakce (CI). Počínaje Hartree–Fockouvou vlnovou funkcí jako determinant základního stavu, vezmeme lineární kombinaci základního stavu a excitované determinanty jako korelované vlnové funkce a optimalizujeme váhové faktory podle variačního principu. Když vezmeme všechny možné excitované determinanty pak mluvíme o Full-CI. Ve Full-CI vlnové funkci jsou všechny elektrony plně korelované. Pro větší molekuly je ale Full-CI příliš výpočetně drahé. Většinou se využívá zkráceného CI rozvoje pro získání dostatečně dobře korelované vlnové funkce a tedy i korelované energie a to podle úrovně zkrácení.


Tato metoda je ve svém jádře podobná CI, ale má několik důležitých výhod proti CI např. je velikostně extenzivní. Meotda vázaných klasrtů využívá tzv. excitační operátory působící na vlnovou funkci například z Hartree–Fockovy metody a to ve tvaru . Takto působící operátor generuje lineární kombinaci Slaterových determinantů, ve kterých jsou elektrony excitovány z okupovaného spinorbitalu do virtuálního. Ořezem excitačního operátoru pak získáváme metodu vázaných klastrů, ve kterých jsou zahrnuty monoexcitace, biexcitace, triexcitace, atd. Metoda vázaných klastrů je velmi široce využívaná[3][4].


- Møller–Plesset poruchová teorie (MP2, MP3, MP4, atd.)
Tyto metody jsou velmi hojně používány v kvantové chemii. Například pro nekovalentní interakce společně s neuronovou sítí dokáže s menší výpočetní náročnosti a přesnosti blížící se k metodě vázaných klastrů, být velmi zajímavou alternativou[5]. Jedná se o poruchovou metodu založenou na předpokladu, že vlnová funkce a energie z HF metody leží blízko exaktnímu řešení a tedy můžeme využít hamiltonián ve tvaru , kde je poruchový parametr (malé reálné číslo) a představuje poruchu. Jelikož energie pro první řád této metody dává energii rovnou původní HF (jelikož ), takže využitelný je až druhý a vyšší řády této metody. Korelační energie se pak dá zapsat jako






- kombinace metod
Využít jde i kombinace různých metod, např. můžeme mít určité téměř degenerované determinanty pro metodu multi-konfiguračního self-konzistentního pole (MCSCF), která bude pro zachycení statické korelace a/nebo CI metodu pro získaní největší části dynamické korelace a/nebo nějaký poruchový ansatz pro malé poruchové (nedůležité) determinanty jako završení. Příklady pro tyto kombinace jsou metody CASPT2 a SORCI.
Matematické hledisko
Pro dva nezávislé elektrony a a b,

kde reprezentuje společnou elektronovou hustotu, nebo hustotu pravděpodobnosti nalezení elektronu v a elektron v . V této notaci, představuje pravděpodobnost nalezení dvou elektronů v jejich příslušných objemových elementech a .








Pokud jsou tyto dva elektrony korelované, pak pravděpodobnost nalezení elektronu v určité poloze v prostoru závisí na poloze elektronu, , a naopak. Jinými slovy, součin jejich nezávislých funkcí hustoty dostatečně nepopisuje skutečnou situaci. Na malých vzdálenostech je hustota nekorelovaných párů příliš velká; na velkých vzdálenostech je hustota nekorelovaných párů příliš malá (tj. elektrony mají tendenci „navzájem se vyhnout“).
Reference
V tomto článku byl použit překlad textu z článku Electronic correlation na anglické Wikipedii.
- LÖWDIN, Per-Olov. Quantum Theory of Many-Particle Systems. III. Extension of the Hartree–Fock Scheme to Include Degenerate Systems and Correlation Effects. Physical Review. American Physical Society, March 1955, s. 1509–1520. DOI 10.1103/PhysRev.97.1509. Bibcode 1955PhRv...97.1509L. (anglicky)
- WIGNER, E. On the Interaction of Electrons in Metals. Physical Review. 1934-12-01, s. 1002–1011. Dostupné online. DOI 10.1103/PhysRev.46.1002. Bibcode 1934PhRv...46.1002W. (anglicky)
- RAMABHADRAN, Raghunath O.; RAGHAVACHARI, Krishnan. Extrapolation to the Gold-Standard in Quantum Chemistry: Computationally Efficient and Accurate CCSD(T) Energies for Large Molecules Using an Automated Thermochemical Hierarchy. S. 3986–3994. Journal of Chemical Theory and Computation [online]. 2013-08-28. Roč. 9, čís. 9, s. 3986–3994. DOI 10.1021/ct400465q.
- ŘEZÁČ, Jan; HOBZA, Pavel. Benchmark Calculations of Interaction Energies in Noncovalent Complexes and Their Applications. S. 5038–5071. Chemical Reviews [online]. 2016-03-04. Roč. 116, čís. 9, s. 5038–5071. DOI 10.1021/acs.chemrev.5b00526.
- MCGIBBON, Robert T.; TAUBE, Andrew G.; DONCHEV, Alexander G.; SIVA, Karthik; HERNÁNDEZ, Felipe; HARGUS, Cory; LAW, Ka-Hei. Improving the accuracy of Møller-Plesset perturbation theory with neural networks. S. 161725. The Journal of Chemical Physics [online]. 2017-10-28. Roč. 147, čís. 16, s. 161725. DOI 10.1063/1.4986081.