Tuberózna skleróza
Tuberózna skleróza je autozomálne dominantne dedičný neurokutánny syndróm (fakomatóza), ktorý sa prejavuje výskytom nezhubných nádorov vo viacerých orgánoch (angiomyolipóm obličky a pečene, rabdomyóm srdca, obrovskobunkový astrocytóm mozgu). Zvyčajne sa diagnostikuje už v ranom detstve.
| Tuberózna skleróza | |
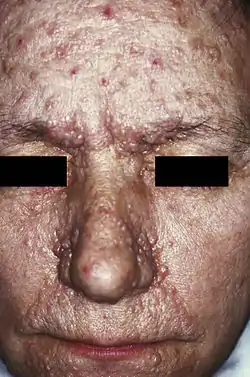 Pacient s adenoma sebaceum, jedným z prejavov tuberóznej sklerózy | |
| Klasifikácia | |
|---|---|
| MKCH-10 | Q85.1 ([ odkaz]) |
| Klinický obraz | |
| Príčina | génová mutácia |
| Postihnutý systém | systémové postihnutie |
|
| |
V roku 1880 ju popísal francúzsky neurológ Désiré-Magloire Bourneville.[1]
Výskyt
Je druhou najčastejšou fakomatózou po neurofibromatóze I. typu, prevalencia sa pohybuje medzi 1: 12 000 až 16 000, celosvetový počet postihnutých sa odhaduje na 1 a pol milióna.
Nie je rozdiel výskytu medzi pohlaviami.
Patogenéza
Príčinou ochorenia sú mutácie na chromozómoch 9 a 16,[2][3] konkrétne dvoch génov:
Oba fungujú ako tumor supresorové gény (ich pôsobky potláčajú rast nádorov).[4]
Mutácie sú autozomálne dominantne dedičné, no približne 2/3 diagnostikovaných prípadov sú sporadické, spontánne mutácie. Približne u 15% prípadov tuberóznej sklerózy sa žiadna z mutácií nenájde; tieto prípady majú miernejší klinický priebeh.
Môže sa vyskytnúť ako samostatná choroba, alebo spojená s ďalšími, ako sú lymfangioleiomyomatóza (takmer výlučne u žien) a autozomálne dominantná polycystická choroba obličiek.
Klinické príznaky a diagnóza
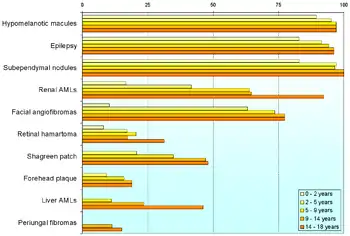
Klasickou „triádou“ príznakov je epilepsia, mentálna retardácia a angiofibrómy tváre (staršie adenoma sebaceum). Kompletne sa však vyskytuje zriedkavo: 50% pacientov nemá poruchu inteligencie (pre postihnutých je typický rôzny stupeň autizmu), 25% nemá epilepsiu. Väčšina pacientov má rôzne prejavy poškodenia centrálnej nervovej sústavy.
Klinické a rádiologické príznaky sa rozdeľujú do dvoch skupín: na hlavné a vedľajšie.[5] Definitívna diagnóza sa stanovuje pri výskyte 2 hlavných, alebo 1 hlavného a dvoch vedľajších príznakov:
- Hlavné príznaky
- časté:
- angiofibrómy tváre - hamartómy zo spojivového a cievneho tkaniva vyskytujúce sa u cca 70% pacientov
- hypomelanotické makuly - viac ako 90% pacientov
- kortikálne tubery
- subependymálne noduly
- bežné:
- retinálny hamartóm
- lymfangioleiomyomatóza - pri poškodení pľúc dýchavica a vykašliavanie krvi
- angiomyolpiómy obličiek - zvyčajne bezpríznakové, no môžu krvácať s klinickými prejavmi bolesti až šoku
- rabdomyóm srdca - obyčajne bez príznakov, no môže spôsobiť arytmiu a náhlu smrť; vekom sa obyčajne postupne zmenšuje
- zriedkavé:
- šagrénové škvrny - hrubé, elastické plôšky (spojivové névy)
- unguálne (nechtové) fibrómy
- subependymálny astrocytóm z obrovských buniek
- Vedľajšie príznaky
- časté:
- mnohopočetné jamky v zubnej sklovine
- rektálne polypy/hamartómy
- bežné:
- kostné cysty
- poruchy migrácie sivej hmoty
- mnohopočetné cysty obličiek
- fibrómy ďasien
- zriedkavé:
- lézie kože podobné konfetám
- achromatické škvrny sietnice
Zobrazovacia diagnostika
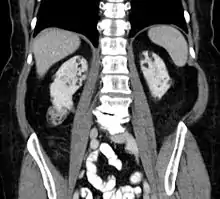
Prvým zobrazovacím vyšetrením pri objavení sa klinických príznakov je MR, prípadne CT mozgu - nález býva pomerne špecifický: viacpočetné kalcifikované subependymálne tubery (histologicky sa jedná o subependymálny astrocytóm z obrovských buniek).
Angiomyolipóm obličky sa u pacientov s tuberóznou sklerózou vyskytuje až v 90% prípadov - počítačová tomografia ho môže diagnostikovať s istotou vtedy, ak v nádore zobrazí tuk (viditeľný u 95% týchto nádorov) a abnormálne cievne štruktúry. V obličkách ďalej môžu byť početné cysty.
Angiomyolipóm sa môže vyskytnúť i v iných orgánoch, najmä v pečeni. Tá môže byť zväčšená a okrem angiomyolipómu sa tu môže objaviť i lipóm, hamartóm či fibróm.
Pankreas môže byť hypoplastický, takisto môže obsahovať hamartómy alebo nádor z ostrovčekovitých buniek.
Pri súčasne sa vyskytujúcej lymfangioleiomyomatóze možno zobraziť hrubé a/alebo tenkostenné cysty v pľúcnom parenchýme a retroperitoneu, zväčšené lymfatické uzliny, ascites a pleurálny výpotok, prípadne rozšírenie ductus thoracicus.
Môžu sa vyskytovať polypy kdekoľvek v priebehu tráviacej trubice.
Sledovanie
Kontrolné ultrasonografické vyšetrenie obličiek každý 1 až 3 roky, v prípade nejasného nálezu alebo zmeny je indikované CT alebo MR.
U dospelých žien doplniť CT vyšetrenie hrudníka na vylúčenie súčasne sa vyskytujúcej lymfangioleiomyomatózy.
Diferenciálna diagnóza
Pri náleze angiomyolipómu obličky treba myslieť na jeho sporadický výskyt - štyria z piatich pacientov s týmto nádorom nemajú tuberóznu sklerózu. Sporadicky sa vyskytujúci angiomyolipóm býva častejší u žien, väčšinou je iba jeden a v jednej obličke.
Autozomálne dominantná polycystická choroba obličiek sa môže vyskytnúť samostatne, ale i v kombinácii s tuberóznou sklerózou. Príčinou je to, že gén TSC2 a gén PKD1 (ktorého mutácia ju spôsobuje) ležia vedľa seba na chromozóme 16.
Komplikácie
- spontánne krvácanie do angiomyolipómu obličky (často riešiteľné iba jej odstránením - nefrektómiou)
- výskyt karcinómu obličky nie je vyšší ako u bežnej populácie, u pacientov s tuberóznou sklerózou sa však objavuje štatisticky v mladšom veku a rastie pomalšie
Prognóza
Prognóza nie je dobrá, 40% pacientov umiera pred dovŕšením 35-ho roku života. Prognóza je horšia najmä pri kombinácii s autozomálne dominantnou polycystickou chorobou obličiek. Príčinami smrti je v dospelosti predovšetkým zlyhanie obličiek, v detstve komplikácie spojené s poškodením mozgu.
Liečba
Liečba je symptomatická: antiepileptická liečba, chirurgické odstránenie symptomatických nádorov a podobne.
Referencie
- Gomez, Manuel R. (apríl 1991), „Phenotypes of the Tuberous Sclerosis Complex with a Revision of Diagnostic Criteria“, Tuberous Sclerosis and Allied Disorders: Clinical, Cellular, and Molecular Studies, Annals of the New York Academy of Sciences, str. 1-7
- The European Chromosome 16 Tuberous Sclerosis Consortium (december 1993), „Identification and characterization of the tuberous sclerosis gene on chromosome 16“ (po anglicky), Cell 75 (7): 1305-1315, ISSN 0092-8674, http://www.sciencedirect.com/science/article/pii/009286749390618Z
- van Slegtenhorst, Marjon; de Hoogt, Ronald (august 1997), „Identification of the Tuberous Sclerosis Gene TSC1 on Chromosome 9q34“ (po anglicky), Science 277 (5327): 805-808, ISSN 0036-8075 (print), 1095-9203 (online), http://www.sciencemag.org/content/277/5327/805.short
- Manning, Brendan D.; Tee, Andrew R. (júl 2002), „Identification of the Tuberous Sclerosis Complex-2 Tumor Suppressor Gene Product Tuberin as a Target of the Phosphoinositide 3-Kinase/Akt Pathway“ (po anglicky), Molecular Cell 10 (1): 151-162, ISSN 1097-2765, http://www.sciencedirect.com/science/article/pii/S1097276502005683
- Roach, E.S.; Gomez, Manuel R; Northrup, Hope (december 1998), „Tuberous Sclerosis Complex Consensus Conference: Revised Clinical Diagnostic Criteria“ (po anglicky), Journal of Child Neurology 13 (12): 624-628, ISSN 0883-0738 (print), 1708-8828 (online), http://jcn.sagepub.com/content/13/12/624.short
Zdroje
- Federle, Michael P.; Jeffrey, R. Brooke; Woodward, Paula J.; Borhani, Amir A. (2010), „Tuberous Sclerosis“ (po anglicky), Diagnostic Imaging Abdomen (2 vyd.), Amirsys Publishing, Inc., str. 8-11, ISBN 978-931884-71-6 Chybné ISBN
- „Tuberous Sclerosis and Allied Disorders: Clinical, Cellular, and Molecular Studies“ (po anglicky), Annals of the New York Academy of Sciences (615): 1-385, apríl 1991, ISSN 1749-6632 (online), http://onlinelibrary.wiley.com/doi/10.1111/nyas.1991.615.issue-1/issuetoc
Ďalšia literatúra
- Gomez, Manuel Rodriguez; Sampson, Julian R.; Whittemore, Vicky Holets (1999) (po anglicky), Tuberous Sclerosis Complex (3 vyd.), New York: Oxford University Press, str. 368, ISBN 0-19-512210-0, http://books.google.sk/books?id=4KeoMozO0wQC&printsec=frontcover&hl=sk#v=onepage&q&f=false