Dokování
Dokování je v oblasti molekulárního modelování in silico metodou, která předpovídá preferovanou orientaci jedné molekuly k druhé při vzájemné vazbě (dochází ke stavbě komplexu). Znalost preferované orientace může být použita k predikci síly asociace molekul, popř. vazebné afinity mezi těmito molekulami. Tuto předpověď lze kvantifikovat pomocí skórovací funkce. Asociace mezi biologicky aktivními molekulami, jako jsou proteiny, peptidy, nukleové kyseliny, sacharidy a lipidy hraje významnou roli především při přenosu signálu. Kromě toho může interakce dvou interagujících partnerů ovlivnit typ produkovaného signálu (agonismus vs. antagonismus). Dokování je tedy důležité i z pohledu predikce produkovaného signálu.[1]
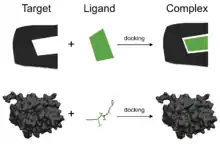
Molekulární dokování je jednou z nejčastěji používaných metod při návrhu nových léčiv založeného na struktuře, a to především díky možnosti predikce vazebné konformace ligandu na vhodné aktivní místo. Charakterizace vazebného chování hraje důležitou roli při racionálním návrhu léčiv, i při objasňování základních biochemických procesů.[2]
Přístupy dokování
V oblasti molekulárního dokování existují dva populární přístupy.
Prvním z nich je shape-complementary docking, který porovnává vztah povrchu molekul ligandu a cílové molekuly.
Druhým z nich je simulation docking, který simuluje skutečný proces dokování a počítá tedy s energetickými interakcemi ligandu a cílové molekuly.[3][4][5]
Mechanismy dokování
Nutnou podmínkou k provedení dokování je znalost cílové molekuly (struktura bývá obvykle stanovena pomocí biofyzikálních metod, jako je rentgenová krystalografie nebo NMR spektroskopie). Známá struktura cílové struktury a databáze potenciálních ligandů pak slouží jako vstupy do dokovacího programu. Úspěšnost dokování pak závisí na dvou komponentech – na užitém algoritmu a skórovací funkci.[6]
Vyhodnocení dokování
Pro správnou predikci vazebných pozic a afinit na cílovou molekulu a pro získání věrohodných dat, je nutné nastavit správné parametry dokování. Pro určení predikčních schopností dokování je obecně vyžadováno posouzení dokovacího protokolu (provádí se porovnáním s experimentálními daty, pokud jsou k dispozici). Posouzení dokování lze provést pomocí různých strategií:[7]
- výpočet přesnosti dokování
- korelace mezi hodnotami získanými ze skórovací funkce a experimentálními výsledky
- pomocí induced-fit modelu.
Odkazy
Reference
- LENGAUER, T.; RAREY, M. Computational methods for biomolecular docking. Current Opinion in Structural Biology. 1996-06, roč. 6, čís. 3, s. 402–406. PMID: 8804827. Dostupné online [cit. 2020-01-07]. ISSN 0959-440X. DOI 10.1016/s0959-440x(96)80061-3. PMID 8804827.
- KITCHEN, Douglas B.; DECORNEZ, Hélène; FURR, John R. Docking and scoring in virtual screening for drug discovery: methods and applications. Nature Reviews. Drug Discovery. 2004-11, roč. 3, čís. 11, s. 935–949. PMID: 15520816. Dostupné online [cit. 2020-01-07]. ISSN 1474-1776. DOI 10.1038/nrd1549. PMID 15520816.
- GOLDMAN, B. B.; WIPKE, W. T. QSD quadratic shape descriptors. 2. Molecular docking using quadratic shape descriptors (QSDock). Proteins. 2000-01-01, roč. 38, čís. 1, s. 79–94. PMID: 10651041. Dostupné online [cit. 2020-01-07]. ISSN 0887-3585. PMID 10651041.
- MENG, Elaine C.; SHOICHET, Brian K.; KUNTZ, Irwin D. Automated docking with grid-based energy evaluation. Journal of Computational Chemistry. 1992-05, roč. 13, čís. 4, s. 505–524. Dostupné online [cit. 2020-01-07]. ISSN 0192-8651. DOI 10.1002/jcc.540130412. (anglicky)
- MORRIS, Garrett M.; GOODSELL, David S.; HALLIDAY, Robert S. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. Journal of Computational Chemistry. 1998, roč. 19, čís. 14, s. 1639–1662. Dostupné online [cit. 2020-01-07]. ISSN 1096-987X. DOI 10.1002/(SICI)1096-987X(19981115)19:143.0.CO;2-B. (anglicky)
- FEIG, Michael; ONUFRIEV, Alexey; LEE, Michael S. Performance comparison of generalized born and Poisson methods in the calculation of electrostatic solvation energies for protein structures. Journal of Computational Chemistry. 2004-01-30, roč. 25, čís. 2, s. 265–284. PMID: 14648625. Dostupné online [cit. 2020-01-07]. ISSN 0192-8651. DOI 10.1002/jcc.10378. PMID 14648625.
- HUANG, Niu; SHOICHET, Brian K.; IRWIN, John J. Benchmarking sets for molecular docking. Journal of Medicinal Chemistry. 2006-11-16, roč. 49, čís. 23, s. 6789–6801. PMID: 17154509 PMCID: PMC3383317. Dostupné online [cit. 2020-01-07]. ISSN 0022-2623. DOI 10.1021/jm0608356. PMID 17154509.