DiGeorgův syndrom
DiGeorgův syndrom (DGS), jinak též velokardiofaciální syndrom či hypoplázie brzlíku a příštitných tělísek) patří mezi takzvané mikrodeleční syndromy či syndromy genů naléhajících na sebe. Syndrom byl popsán v roce 1968 dětským endokrinologem Angelo DiGeorgem.[1][2]
| DiGeorgův syndrom | |
|---|---|
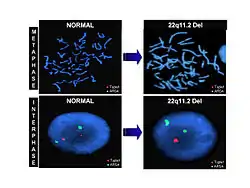 FISH analýza pacienta s DG | |
| Klasifikace | |
| MKN-10 | D82.1 |
| Některá data mohou pocházet z datové položky. | |
Etiologie a patogeneze
Nejčastější příčinou tohoto syndromu je delece na dlouhém raménku 22. chromozomu (úsek 22q11), která je přítomná u 90 % pacientů s DGS. Stejnou deleci nacházíme i u dvou dalších, fenotypově téměř totožných syndromů – Takaova syndromu a Shprintzenova syndromu. Pro samotný DiGeorgův syndrom je pak nejtypičtější delece úseku 22q11.2. Popsány byly případy s mutací T-BOX 1 genu (TBX1) – tedy specifického transkripčního faktoru.
Syndrom se vyskytuje většinou sporadicky, popsány jsou však i případy familiárního výskytu (výskyt v rodině), kde syndrom vykazoval autozomálně dominantní typ dědičnosti. Mimo vlastní deleci 22q11 byly popsány i translokace – např. t(2; 22), t(4; 22) či t(20; 22). Charakteristické příznaky tohoto syndromu byly popsány i u delecí na jiných chromosomech, než v oblasti 22q11 – například del(10p13), del(18q21.33) či del(4q21.3-q25).
Vzhledem k podobným fenotypovým projevům (obecně velokardiofaciální anomálie) a stejné deleci jsou v poslední době DiGeorgův, Shprintzenův a Takaův syndrom řazeny pod označení CATCH 22 (anglicky: Cardiac abnormality / abnormal facies (vrozené srdeční vady), T cell deficit due to thymic hypoplasia (hypoplázie či ageneze thymu vedoucí k imunodeficienci), Cleft palate (rozštěp patra), Hypocalcemia due to hypoparathyroidism resulting from 22q11 deletion (hypoparathyroidismus vedoucí k hypokalcémii); akronym CATCH 22 je inspirován anglickým názvem knihy Hlava XXII (Catch-22) od Josepha Hellera.
Klinický obraz
DiGeorgův syndrom je porucha vývoje třetí a čtvrté žaberní výchlipky, jejímž následkem je omezený vývoj (až úplná absence) brzlíku a příštitných tělísek. Narušen může být i vývoj štítné žlázy a ultimobranchiálního tělíska. Typické jsou rovněž vrozené vady srdce a velkých cév a různé abnormality v obličejové krajině včetně rozštěpů. Relativně častá je i mentální retardace.
Projevy jsou značně proměnlivé a závažnost postižení je úměrná deficitu T-lymfocytů, ty jsou přítomny pouze v nízkých hladinách a v některých případech zcela chybí. Redukovány jsou i orgánové thymodependentní oblasti jako parakortikální zóny lymfatických uzlin. Deficit T-lymfocytů má sklon se s věkem normalizovat a okolo 5 let věku mohou T-lymfocyty dosáhnout normálních hodnot. Mimo poruchy imunity a náchylnosti především k některým virovým a mykotickým infekcím se syndrom vyznačuje i hypokalcémií (kvůli nepřítomnosti parathormonu z příštitných tělísek) a případnou tetanií.
Terapie
Léčba je symptomatická. V těžších případech je možnost transplantace kostní dřeně s periferními lymfocyty či transplantát z kultivované thymové tkáně. Obličejové a srdeční vady je možno řešit chirurgickou cestou.
Odkazy
Reference
- V tomto článku je použit text článku DiGeorgův syndrom ve WikiSkriptech českých a slovenských lékařských fakult zapojených v MEFANETu.
- DiGeorge AM. Congenital absence of the thymus and its immunologic consequences: concurrence with congenital hypoparathyroidism. IV(1). White Plains, NY: March of Dimes-Birth Defects Foundation; 1968:116-21
- RESTIVO, Angelo; SARKOZY, Anna; DIGILIO, Maria Cristina; DALLAPICCOLA, Bruno; MARINO, Bruno. 22q11 Deletion syndrome: a review of some developmental biology aspects of the cardiovascular system.. Journal of Cardiovascular Medicine. February 2006, roč. 7, čís. 2, s. 77–85. Dostupné online. DOI 10.2459/01.JCM.0000203848.90267.3e.
Literatura
- BARTŮŇKOVÁ, Jiřina. Imunodeficience. 1. vyd. Praha: Grada, 2002. ISBN 80-247-0244-4.
Externí odkazy
 Obrázky, zvuky či videa k tématu DiGeorgův syndrom na Wikimedia Commons
Obrázky, zvuky či videa k tématu DiGeorgův syndrom na Wikimedia Commons - ŠÍPEK, Antonín. Geneticky podmíněné poruchy imunitního systému [online]. 2006-06-09 [cit. 2010-09-16]. Dostupné v archivu pořízeném dne 2010-07-22.