Strukturní alignment
Strukturní alignment je způsob porovnávání polymerních struktur na základě jejich tvaru a prostorového rozložení. Kromě určování podobností proteinů nachází využití i při porovnávání molekul RNA.
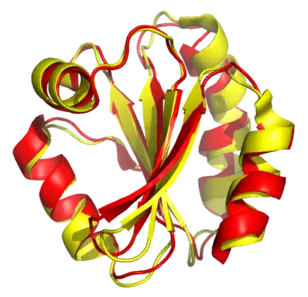
Určení podobnosti proteinů patří mezi důležité faktory správné klasifikace proteinů, ze strukturní podobnosti s ostatními proteiny lze vyvodit informace o evolučním vývoji, nebo biologické funkci, čehož se využívá např. při klasifikaci nově objevených proteinů, u kterých je známa pouze struktura. Pro strukturní alignment není potřeba předem znát informace o společných oblastech proteinů. V případě, že mají proteiny velkou sekvenční podobnost, využívá se spíše sekvenční alignment namísto strukturního.
Pro tvorbu alignmentu existují desítky algoritmů, liší se především v reprezentaci terciární struktury a rozdílnému přístupu ke kvantifikaci rozdílů ve struktuře. Optimálním alignmentem nazýváme soustavy souřadnic v prostoru reprezentující porovnávané proteiny, které se překrývají tak, aby mezi nimi byla nejmenší možná odchylka.
Ohodnocení podobnosti proteinů
Tvorbu alignmentu předchází analýza porovnávaných proteinů, za účelem zjištění podobností jednotlivých oblastí. K tomu je potřeba vytvořit takovou počítačovou reprezentaci proteinů, aby umožnila výpočet. Obecně je lze rozdělit na trojrozměrnou (3D), dvojrozměrnou (2D), jednorozměrnou (1D), popř. lze charakterizovat protein jedinou hodnotou (0D). Ačkoli redukcí struktury ztrácíme informace, zvyšujeme tím rychlost algoritmu a v některých případech je to žádoucí. Problém nalezení všech podobných oblastí proteinů totiž patří do třídy NP-těžkých úloh a pouze pomocí správné reprezentace dat a funkce pro ohodnocení porovnání lze zkonstruovat algoritmus řešící problém v rozumném výpočetním čase.[1]
3D reprezentace
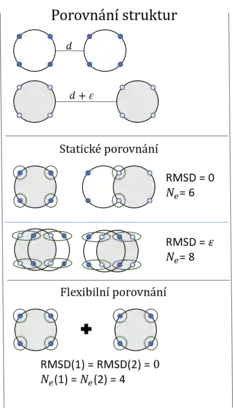
U 3D reprezentace se měří rozdíly v pozici ekvivalentních atomů, buď při pevně daném přiřazení k sobě (statické porovnání), nebo při možnosti natočení určitých oblastí (flexibilní porovnání). Flexibilní způsob umožňuje otočit určité úseky v rámci molekul blíže k sobě, čímž je umožněno určit podobnost mezi proteiny s konformační odlišností. Díky tomu podává přesnější informace a je v současnosti nejvíce využíván.[2][3][4][5][6] Samotné hodnocení podobnosti závisí na několika faktorech – rozdílu v počtu společných domén (), rozdílu v pozicích atomů (root mean square deviation - ) a v penalizování mezer.[1]


2D reprezentace
Zjednodušení struktury pomocí ekvivalence reziduí, na základě totožných terciárních interakcí. Interakce mezi rezidui je popsána pomocí kontaktních map, grafů, nebo vzdálenostních matic. Využívá se topologické analýzy proteinů.[7][8][9] Oproti 3D reprezentacím nejsou vyhodnocovány vzdálenosti mezi terciárními strukturami, ale pouze mezi páteří jednotlivých proteinů.[1]
1D reprezentace
1D reprezentaci se také říká strukturní profil. Jedná se o klasifikaci každého rezidua na základě typu aminokyseliny a konformačním stavu páteře. Celý protein je zredukován do řetězce symbolů, na který lze jednoduše aplikovat optimalizované algoritmy,[10][11][9] avšak taková redukce sebou nese limitované možnosti detekce podobnosti proteinů, které mají rozdílnou nadstavbu na základní kostře.
0D reprezentace
Redukce celého proteinu do jedné hodnoty, popřípadě histogramu.[12][13] Podobné struktury generují stejné, nebo přibližně stejné hodnoty. Uložení v tabulkách (databázích) a nahlížení na hodnoty pomocí indexu poskytuje nejrychlejší způsob hledání. Problémem je nemožnost porovnání podoblastí proteinů.[1]
Algoritmy pro alignment
Jakmile je známa podobnost proteinů, je zkonstruován optimální alignment. Algoritmy klasicky využívají různé heuristické přístupy, aby zjednodušili výpočet.[1]
Combinatorial extension (CE)
Algoritmus CE byl vymyšlen již v roce 1998. Algoritmus nejprve rozdělí proteiny na fragmenty, ze kterých vytvoří sub alignmenty AFP (z angl. Alignment Fragment Pairs). Postupných přidáváním dalších párů dochází k vytvoření úplného alignmentu. Délka AFP je v ideálním případě 8 aminokyselin. Uživatel si však může ovlivňovat konečný výsledek pomocí volby parametrů, dle kterých bude algoritmus pracovat. Kromě délky AFP to může být třeba délka možné mezery mezi jednotlivými AFP. CE je využíván např. v databázi PDB pro porovnávání proteinů.[14]
Distance alignment matrix method (DALI)
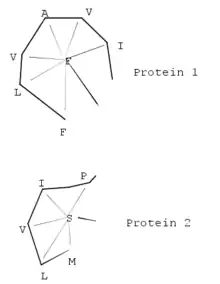
Podobně jako CE je založen na fragmentaci struktury proteiny, ale ideálně na hexapeptidové úseky. Následně vytvoří vzdálenostní matici vyhodnocením kontaktů mezi fragmenty jednotlivých proteinů. Na hlavní diagonále jsou obsaženy rezidua, které tvoří určitou sekundární struktury a nacházejí se vedle sebe v sekvenci. Na vedlejších diagonálách jsou reprezentovány úseky, které se prostorově dotýkají, ale nenacházejí se vedle sebe v sekvenci. Pokud jsou diagonály rovnoběžné, jedná se o typ sekundární struktury, která se vyskytuje paralelně. V případě, že jsou diagonály kolmé, jsou sekundární struktury antiparalelní. Taková matice je vytvořena pro každý protein a dále jsou analyzovány jejich podobnosti.[15] DALI je možné využívat na webu.
Sequential structure alignment program (SSAP)
Využívá dvojité dynamické programování k vytvoření alignmentu. Na rozdíl od většiny algoritmů nepracuje s alfa uhlíky pro lokalizaci aminokyselin, ale s beta uhlíky. Tímto je umožněno při porovnávání zahrnout rotaci daného rezidua. Nejprve dojde ke konstrukci vzdálenostních vektorů mezi každým reziduem a jeho nejbližším sousedem, který se nenachází hned vedle v sekvenci. Z vektorů je následně vytvořena matice, ze které je pomocí dynamického programování získán ideální alignment. Původně byl algoritmus využit pouze pro párový alignment, ale je již optimalizován i pro vícenásobný.[16] Je využíván např. v databázi CATH.
RNA strukturní alignment
Dlouhé RNA molekuly tvoří charakteristické terciární struktury, které jsou udržovány vodíkovými vazbami mezi nukleotidovými bázemi. Alignment nekódujících RNA molekul je důležitý, protože struktura je více konzervovaná než sekvence bází.[17] Příkladem softwaru, sloužícímu k RNA alignmentu je SETTER, který pomocí alignmentu fragmentů RNA vytvoří optimální celkový alignment. Lze jej využívat skrze webové stranky.
Reference
- HASEGAWA, Hitomi; HOLM, Liisa. Advances and pitfalls of protein structural alignment. Current Opinion in Structural Biology. 2009-06, roč. 19, čís. 3, s. 341–348. Dostupné online [cit. 2020-01-28]. ISSN 0959-440X. DOI 10.1016/j.sbi.2009.04.003.
- CSABA, G.; BIRZELE, F.; ZIMMER, R. Protein structure alignment considering phenotypic plasticity. Bioinformatics. 2008-08-09, roč. 24, čís. 16, s. i98–i104. Dostupné online [cit. 2020-01-28]. ISSN 1367-4803. DOI 10.1093/bioinformatics/btn271.
- GUERLER, Aysam; KNAPP, Ernst-Walter. Novel protein folds and their nonsequential structural analogs. Protein Science. 2008-08, roč. 17, čís. 8, s. 1374–1382. Dostupné online [cit. 2020-01-28]. ISSN 0961-8368. DOI 10.1110/ps.035469.108.
- MARTÍNEZ, Leandro; ANDREANI, Roberto; MARTÍNEZ, José Mario. Convergent algorithms for protein structural alignment. BMC Bioinformatics. 2007-08-22, roč. 8, čís. 1. Dostupné online [cit. 2020-01-28]. ISSN 1471-2105. DOI 10.1186/1471-2105-8-306.
- MENKE, Matthew; BERGER, Bonnie; COWEN, Lenore. Matt: Local Flexibility Aids Protein Multiple Structure Alignment. PLoS Computational Biology. 2008, roč. 4, čís. 1, s. e10. Dostupné online [cit. 2020-01-28]. ISSN 1553-734X. DOI 10.1371/journal.pcbi.0040010.
- MOSCA, R.; SCHNEIDER, T. R. RAPIDO: a web server for the alignment of protein structures in the presence of conformational changes. Nucleic Acids Research. 2008-05-19, roč. 36, čís. Web Server, s. W42–W46. Dostupné online [cit. 2020-01-28]. ISSN 0305-1048. DOI 10.1093/nar/gkn197.
- BIRZELE, F.; GEWEHR, J. E.; CSABA, G. Vorolign--fast structural alignment using Voronoi contacts. Bioinformatics. 2007-01-15, roč. 23, čís. 2, s. e205–e211. Dostupné online [cit. 2020-01-28]. ISSN 1367-4803. DOI 10.1093/bioinformatics/btl294.
- LESLIN, C. M.; ABYZOV, A.; ILYIN, V. A. TOPOFIT-DB, a database of protein structural alignments based on the TOPOFIT method. Nucleic Acids Research. 2007-01-03, roč. 35, čís. Database, s. D317–D321. Dostupné online [cit. 2020-01-28]. ISSN 0305-1048. DOI 10.1093/nar/gkl809.
- SACAN, A.; TOROSLU, I. H.; FERHATOSMANOGLU, H. Integrated search and alignment of protein structures. Bioinformatics. 2008-10-22, roč. 24, čís. 24, s. 2872–2879. Dostupné online [cit. 2020-01-28]. ISSN 1367-4803. DOI 10.1093/bioinformatics/btn545.
- LIU, Xin; ZHAO, Ya-Pu; ZHENG, Wei-Mou. CLEMAPS: Multiple alignment of protein structures based on conformational letters. Proteins: Structure, Function, and Bioinformatics. 2008-05-01, roč. 71, čís. 2, s. 728–736. Dostupné online [cit. 2020-01-28]. ISSN 0887-3585. DOI 10.1002/prot.21739.
- SIERK, M. L. Sensitivity and selectivity in protein structure comparison. Protein Science. 2004-02-06, roč. 13, čís. 3, s. 773–785. Dostupné online [cit. 2020-01-28]. ISSN 0961-8368. DOI 10.1110/ps.03328504.
- CHU, Chia-Han; TANG, Chuan Yi; TANG, Cheng-Yin. Angle-distance image matching techniques for protein structure comparison. Journal of Molecular Recognition. 2008-11, roč. 21, čís. 6, s. 442–452. Dostupné online [cit. 2020-01-28]. ISSN 0952-3499. DOI 10.1002/jmr.914.
- KONAGURTHU, Arun S.; STUCKEY, Peter J.; LESK, Arthur M. Structural search and retrieval using a tableau representation of protein folding patterns. Bioinformatics. 2008-01-05, roč. 24, čís. 5, s. 645–651. Dostupné online [cit. 2020-01-28]. ISSN 1460-2059. DOI 10.1093/bioinformatics/btm641.
- SHINDYALOV, I. N.; BOURNE, P. E. Protein structure alignment by incremental combinatorial extension (CE) of the optimal path. Protein Engineering Design and Selection. 1998-09-01, roč. 11, čís. 9, s. 739–747. Dostupné online [cit. 2020-01-28]. ISSN 1741-0126. DOI 10.1093/protein/11.9.739.
- HOLM, Liisa; SANDER, Chris. Dali: a network tool for protein structure comparison. Trends in Biochemical Sciences. 1995-11, roč. 20, čís. 11, s. 478–480. Dostupné online [cit. 2020-01-28]. ISSN 0968-0004. DOI 10.1016/s0968-0004(00)89105-7.
- SHATSKY, Maxim; NUSSINOV, Ruth; WOLFSON, Haim J. Algorithms for Multiple Protein Structure Alignment and Structure-Derived Multiple Sequence Alignment. New Jersey: Humana Press Dostupné online. ISBN 1-59745-574-1. S. 125–146.
- TORARINSSON, E. Thousands of corresponding human and mouse genomic regions unalignable in primary sequence contain common RNA structure. Genome Research. 2006-07-01, roč. 16, čís. 7, s. 885–889. Dostupné online [cit. 2020-01-28]. ISSN 1088-9051. DOI 10.1101/gr.5226606.