Cri du chat
Cri du chat (též syndrom kočičího pláče/mňoukání/křiku či Lejeunův syndrom, zapisován „46, XX, del(5p)“) je vzácná vrozená geneticky podmíněná choroba, kterou v roce 1963 objevil a popsal francouzský lékař a genetik Jérôme Lejeune. Vyskytuje se velmi vzácně (asi jeden případ na 20–50 tisíc živých porodů).
| Cri du chat | |
|---|---|
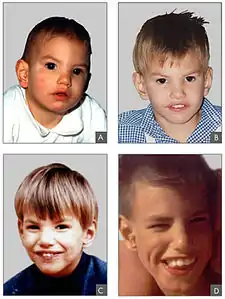 Pacient postižený syndromem Cri du chat ve věku: A) 8 měsíců, B) 2 let, C) 4 let a D) 9 let | |
| Klasifikace | |
| MKN-10 | Q93.4 |
| Statistické údaje – obě pohlaví[1][2][3][4][5] | |
| Prevalence | 1 / 50 000 živě narozených |
| Některá data mohou pocházet z datové položky. | |
Genetické podklady
Příčinou choroby je úplná či částečná ztráta (delece) krátkého raménka 5. chromozomu, která přibližně v 80 % vzniká de novo (tj. bez předchozího nálezu v rodině nebo ve formě balancované přestavby u jednoho z rodičů). Za příčinu typických projevů byla konkrétně identifikována delece kritického regionu umístěného v úseku 5p15.2. Do této oblasti byly mapovány geny pro proteiny Semaforin F a δ-catenin, jež mají zřejmě funkci v nervovém systému.
Kritický lokus se nalézá v subtelomerické oblasti krátkého raménka pátého chromozomu, a proto Cri du chat syndrom bývá zařazován mezi tzv. subtelomerické aberace. Zároveň, pokud se jedná o deleci takto malého rozsahu, jež není rozlišitelná základní cytogenetickou analýzou (metodou G-pruhování), bývá tento syndrom řazen i mezi tzv. mikrodeleční syndromy.
Nejčastější příznaky a projevy
Hlavním projevem u většiny novorozenců, podle něhož syndrom nese své jméno, je pláč podobný kočičímu mňoukání, který může vyplývat z anatomicky nesprávně utvářeného hrtanu a hrtanové příklopky. Novorozenec trpí hypotonií, má nízkou porodní váhu, navíc často v kombinaci s potížemi sání a polykání. Z tohoto důvodu může dítě v prvních dvou letech hůře prospívat. Mimoto jsou časté i dýchací obtíže komplikované respiračními infekcemi. V novorozeneckém a kojeneckém věku bývá popisována mikrocefalie a tzv. "měsíčkovitý" obličej, jenž se během stárnutí stává méně zřetelným. Dospělý má spíše protáhlý obličej s typickými projevy – epikanty, širokým kořenem nosu, krátkým philtrem a ústy s "dolu stočenými" koutky. U postiženého se rozvíjí velmi těžká psychomotorická a mentální retardace. Chůze se rozvíjí až mezi 5. a 6. rokem života. Komunikační schopnosti, pokud se vyvinou, jsou značně omezené, přičemž vhodnou alternativou může být znaková řeč. Často se přidružuje hyperaktivita a ztráty pozornosti. Po 20. roce se u postižených objevují presenilní projevy, jež se postupem času prohlubují.
Prognóza
Prognóza pacientů do jisté míry závisí na včasnosti diagnózy a zahájení terapeutických a preventivních metod zaměřených především na rozvoj mentálních a motorických dovedností jedince.
Stanovení diagnózy
V některých případech může být diagnóza stanovena již během prenatálních vyšetření. A to především na podkladě familiárního výskytu balancované přestavby a předešlého narození postiženého jedince, nicméně opakovaný výskyt de novo mutace je málo pravděpodobný. K vlastnímu stanovení diagnózy často bývá dostačující konvenční cytogenetické vyšetření, případně doplněné o metodu fluorescenční in situ hybridizace s užitím lokus specifické sondy k danému kritickému regionu.
Reference
- CHEN, Harold. Cri-du-chat Syndrome [online]. eMedicine Pediatrics [cit. 2009-08-25]. Dostupné online. (anglicky)
- RODRÍGUEZ-CABALLERO, Á. et al. Cri du chat syndrome: A critical review. [online]. Med Oral Patol Oral Cir Bucal. 3: 473-478. [cit. 2013-01-31]. Dostupné online. (anglicky)
- CERRUTI MAINARDI, P. Cri du Chat syndrome. [online]. Orphanet Journal of Rare Diseases. [cit. 2013-01-31]. Dostupné online. (anglicky)
- TEOH, X. H. Cri-du-chat Syndrome [online]. Singapore Med J [cit. 2013-01-31]. Dostupné online. (anglicky)
- SEEMANOVÁ, E. Mikrodeleční syndromy. [online]. Čas. Lék. čes. [cit. 2013-01-31]. Dostupné online. (česky)
Externí odkazy
 Obrázky, zvuky či videa k tématu Cri du chat na Wikimedia Commons
Obrázky, zvuky či videa k tématu Cri du chat na Wikimedia Commons
Přečtěte si prosím pokyny pro využití článků o zdravotnictví.